
Abstract
Cerebral ischemia causes neuronal death and disruption of neural circuits in the central nervous system. Various neurological disorders caused by cerebral infarction can severely impair quality of life and are potentially fatal. Functional recovery in the chronic stage mainly depends on physical treatment and rehabilitation. We aim to establish cell therapy for cerebral ischemia using embryonic stem (ES) cells, which have self-renewing and pluripotent capacities. We previously reported that the transplanted monkey and mouse ES cell-derived neural progenitors, by stromal cell-derived inducing activity method, could survive and differentiate into various types of neurons and glial cells, and form the neuronal network in basal ganglia. In this report, we induced the differentiation of the neural progenitors from mouse ES cells using the serum-free suspension culture method and confirmed the expression of various basal ganglial neuronal markers and neurotransmitter-related markers both in vitro and in vivo, which was thought to be suitable for replacing damaged striatum after middle cerebral artery occlusion. This is the first report that used selectively induced telencephalic neural progenitors into ischemia model. Furthermore, we purified the progenitors expressing the neural progenitor marker Sox1 by fluorescence-activated cell sorting and Sox1-positive neural progenitors prevented tumor formation in ischemic brain for 2 months. We also analyzed survival and differentiation of transplanted cells and functional recovery from ischemic damage.
Similar content being viewed by others


Main
Regenerative therapy for cerebral ischemia is divided into two approaches: activating endogenous neural stem cells and transplantation of neurons or other cells including neural stem cells.1, 2 Neural stem cells can not only provide trophic factors to support cellular survival and function, but also replace dead neurons and repair damaged neural circuits.3 Even in the adult central nervous system (CNS), endogenous neurogenesis is enhanced after ischemic injury and has a limited capacity to replace lost neurons.4 In comparison, several stem-cell-based transplantation studies for cerebral ischemia have demonstrated safety and functional recovery.5, 6, 7, 8, 9 Transplanted stem cells have been isolated from various sources, such as blastocysts, fetal and adult CNS, bone marrow, and umbilical cord. Among them, embryonic stem (ES) cells are self-renewing, pluripotent cells derived from the inner cell mass of the pre-implantation blastocyst. These cells have many characteristics required of a cell source for cell replacement therapy, including proliferation and differentiation.10 Embryonic neural progenitors survive longer than adult neural progenitors, with less inflammation.8 Buhnemann et al11 showed that transplanted ES cells demonstrate characteristics of electrophysiologically functional neurons with voltage-gated sodium currents. We previously reported that transplanted monkey and mouse ES cells could survive and differentiate into various types of neurons and glial cells, and reconstruct the neuronal network in mouse basal ganglia after middle cerebral artery (MCA) occlusion.12, 13
To establish ES cell-based therapy for clinical applications, it is indispensable to conduct proper cell induction, prevent teratoma formation, and evaluate functional recovery in an animal model. In this report, we transplanted the neural progenitors derived from ES cells into the ischemic mouse putamen after transient MCA occlusion. We inspired differentiation of the neural progenitors from mouse ES cells into the various striatum neurons using the serum-free suspension culture (SFEB culture) method. SFEB culture can particularly induce telencephalic progenitors from floating aggregates of ES cells.14 Following MCA occlusion, primarily telencephalic neurons are lost. Thus, a supply of telencephalic neurons into the brain after MCA occlusion is desired.
The pluripotency of undifferentiated ES cells could result in teratoma formation,15, 16 and we previously demonstrated that purification of the progenitors expressing the neural progenitor marker Sox1 by fluorescence-activated cell sorting (FACS) could prevent tumor formation in healthy mice.17 However, the behavior of neural progenitors sorted by Sox1 in ischemic brain has not been clarified. This is the first report that shows SFEB culture could induce the striatum neurons from ES cells effectively both in vitro and in vivo and Sox1-based cell sorting of neural progenitors prevented tumor formation in ischemic brain for 8 weeks. In addition, we verified neuronal network formation and functional recovery from ischemic damage.
MATERIALS AND METHODS
Maintenance of Mouse ES Cells
Mouse Sox1-GFP knock-in ES cells (46C) were generously provided by Dr Austin Smith (University of Edinburgh, Edinburgh, UK; http://www.ed.ac.uk). The generation of 46C cells has been described previously.18 For the following transplantation experiments, the generated 46C cells expressing β-galactocerebroside (β-gal), selected by G418 (200 mg/ml; Sigma, St Louis, MO, USA; http://www.sigmaaldrich.com), were used in order to identify the transplanted cells, because after transplantation Sox1-GFP ES cells would fade GFP expression with their differentiation.17 All recombinant DNA research conformed to National Institutes of Health (NIH) guidelines.
Undifferentiated 46C cells were maintained on gelatin-coated dishes in G-MEM (Gibco-Invitrogen, Carlsbad, CA, USA) supplemented with 1% fetal calf serum (JRH Biosciences, Lenexa, KS, USA), 8% KSR (knockout serum replacement) (Gibco-Invitrogen, Tokyo, Japan), 2 mM glutamine, 0.1 mM non-essential amino acids, 1 mM pyruvate, 0.1 mM 2-mercaptoethanol (2-ME), and 2000 U/ml leukemia inhibitory factor (Chemicon International, Temecula, CA, USA). For the stromal cell-derived inducing activity (SDIA) culture, ES cells were dissociated (0.25% trypsin-EDTA) to single cells, and 1 × 102 cells were plated on PA6 cells as described19 and cultured in differentiation medium for 21 days. In SFEB culture, ES cell aggregates were replated en bloc in dishes coated with laminin at a density of l × l02 to 2 × l07 aggregates per cm2, on differentiation day 4 of SFEB culture with differentiation medium, and cultured for a further 17 days.14 For both SDIA and SFEB cultures, differentiation medium was prepared as follows: α-MEM (Gibco-Invitrogen) supplemented with 8% KSR (Gibco-Invitrogen), 2 mM glutamine, 1 mM pyruvate, 0.1 mM non-essential amino acids, and 0.1 mM 2-ME. The day on which ES cells were seeded to differentiate was defined as differentiation day 0.
FACS
ES cell colonies differentiated in the SFEB culture for 4 days were isolated using 0.25% trypsin-EDTA, dissociated into a single-cell suspension, and resuspended in cold differentiation medium. To separate two distinct cell populations, Sox1-GFP-positive and Sox1-GFP-negative cells were sorted using a FACSAria cell sorter and FACSDiva software (Beckton Dickinson, Franklin Lakes, NJ, USA) as described previously.17 Dead ES cells were identified and eliminated by propidium iodide staining and forward-side scatter gating, respectively. Gates for each population were set so that the two subsets sorted based on Sox1 staining would not overlap when reanalyzed. Sorted cells were immediately either transplanted or replated onto chamber slides to characterize their behavior in vitro.
Immunohistochemistry
After fixation in 4% paraformaldehyde in PBS (PFA), immunohistochemical stainings were performed with primary antibodies for overnight at 4 °C. The sections were then incubated for 1 h at room temperature with appropriate secondary antibodies (1:400; Molecular Probes, Eugene, OR, USA). Primary antibodies used were mouse anti-NeuN (1:200; Chemicon, Billerica, MA, USA), mouse anti-neuronal class III β-tubulin (TuJ1, 1:500; Covance, Princeton, NJ, USA), rabbit anti-glutamate decarboxylase (GAD, 1:100; Chemicon), rabbit anti-choline acetyltransferase (ChAT, 1:100; Chemicon), rabbit anti-tyrosine hydroxylase (TH, 1:60; Chemicon), rat anti-serotonin (1:100; Chemicon), rabbit anti-calbindin (1:500; Chemicon), rabbit anti-DARPP-32 (1:500; Chemicon), mouse anti-parvalbumin (1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse anti-synaptophysin (1:200; Chemicon), anti-GFP (mouse, 1:200; rabbit, 1:500; Chemicon), rabbit anti-Fluorogold (FG, 1:5000; Chemicon), rabbit anti-Ki67 (1:200; Novocastra, Wetzlar, Germany), and rabbit anti-Oct4 (1:5000; Santa Cruz Biotechnology). Photomicrographs were taken with an Olympus confocal laser scanning microscope (FV-300; Olympus, Tokyo, Japan).
Transient Focal Cerebral Ischemia
All animal experiments were approved and carried out in accordance with the Institutional Animal Care and Use Committee of Kyoto University. C57BL/6 male mice weighing 25–30 g were used.
Cerebral ischemia was induced by using the standard intraluminal MCA occlusion method as described previously.20 Briefly, anesthesia was induced in the mice by administration of 3% halothane in 30% oxygen and 70% nitrous oxide, and maintained using a mixture containing 1% halothane. Rectal temperature was maintained at 37.0±0.5 °C with a thermostat-controlled heating pad. Left MCA was occluded by a 8.0 nylon monofilament coated with silicon. After 45 min, the filament was withdrawn and the mouse was placed in a thermally controlled incubator (32.0 °C) for 2 h. Regional cerebral blood flow was measured with a laser-Doppler flowmetry (Omegawave, Tokyo, Japan) to confirm induction of ischemia. The surgical procedure was considered successful at >80% reduction from base line.
Transplantation
At 7 days after ischemia, the mice were placed in a stereotaxic frame (Narishige, Tokyo, Japan) under deep anesthesia with sodium pentobarbital (40 mg/kg). Each animal received an injection of 2 μl (1 μl/min, 1 × 105 cells per μl) of one of two FACS-sorted cell populations, Sox1-GFP+ and Sox1-GFP− cells, or differentiation medium alone into the lesioned striatum (from bregma: A, −0.5 mm; L, +2.0 mm; V, +3.0 mm; incisor bar, 0) via a Hamilton microsyringe fitted with a 26-G blunt needle.
At 2 months after transplantation, mice were transcardially perfused first with PBS, and then with 4% PFA under deep anesthesia. Brains were post-fixed for 12 h in 4% PFA, and then frozen in Tissue-Tek optimal cutting temperature (OCT) compound (Sakura Finetechnical, Tokyo, Japan) and cut at 40 μm thickness. The sections were processed for immunohistochemistry, as described above.
Retrograde Tracing
The kinetics of transplanted neural progenitors was evaluated using retrograde tracer, hydroxystilbamidine (2% solution in saline; equivalent to FG) (Molecular Probes).13 Briefly, at 7 days before perfusion, the mice were anesthetized with 2% halothane and then received an injection of 0.2 μl (0.05 μl/min) of hydroxystilbamidine into the substantia nigra (from bregma: A, +3.0 mm; L, +1.5 mm; V, +4.25 mm; incisor bar 0) on the grafted side.
Graft and Teratoma Formation Volume
Graft cells were identified by β-gal or GFP immunoreactivity. The graft area was delineated and quantified using Image Pro Plus 6.0, and graft volume was calculated by summing the graft area over every 10th 40-μm-thick slice. The observation of a Ki67-positive cell mass was defined as positive tumor formation.17 Teratoma volume was evaluated in the same way as graft size.
Neurological Evaluation
Neurological evaluation was performed before, and every 7 days after, transplantation. The body swing test was used to evaluate asymmetric motor behavior, as described previously.21 Ischemic mice show a tendency to turn toward the contralateral side after hanging upside down. We assessed the number of right side turns over 10 trials. The corner test was performed to detect long-term sensorimotor dysfunction as described previously.22 Briefly, a mouse was placed between two boards attached at 30° to encourage entry into the corner. The mouse reached the deep part of the corner, and then reared and turned back either on the right or left. Ischemic mice preferentially turn toward the non-impaired, ipsilateral (left) side. The number of left turns was recorded from 10 trials.
Statistical Analysis
Quantitative data were expressed as mean±s.d. Statistical comparisons were conducted using the Wilcoxon Mann–Whitney test for two independent samples, and Kruskal–Wallis test for the comparison among three groups. P-values of <0.05 were considered statistically significant.
RESULTS
Effective Induction to Striatum Neurons
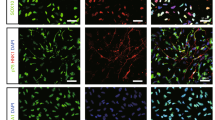
Pax2 is a transcription factor critically required during development of the midbrain and caudal CNS.23 The number of Pax2-positive cells in SFEB culture at day 10 was decreased when compared with SDIA culture (P=0.029). After further culture on laminin-coated dishes, telencephalic progenitors (Bf-1-positive cells) were generated (Figure 1a). Differentiation of the induced neurons was not sufficient at 14 days, but after 21 days, striatum neuron markers were detected: DARRP32, 26.0±10.2; GAD65, 23.2±7.7; calbindin, 9.7±6.6; glutamate, 9.1±4.2; serotonin, 8.7±4.7; TH, 3.7±2.2; ChAT, 2.7±2.1; and parvalbumin, 1.2±1.1 (Figures 1b–i). DARRP32- and GAD65-positive neurons were significantly increased in SFEB culture (P<0.001), in contrast with the increase of TH-positive neurons in SDIA culture, suggesting that SFEB culture promoted telencephalic differentiation and specifically induced mature neurons of basal ganglia (P=0.029) (Figures 1j and k).


Induction of mature striatum neurons derived from embryonic stem (ES) cells, cultured for 4 days in serum-free suspension culture (SFEB culture) and a further 17 days in differentiation medium. Immunohistochemistry of SFEB indicated neural progenitors expressing the telencephalic marker, Bf-1 (a), and striatum neuron-related markers, DARRP32, glutamate decarboxylase (GAD)65, calbindin, glutamate, serotonin, tyrosine hydroxylase (TH), choline acetyltransferase (ChAT), and parvalbumin (b–i). Scale bar: 100 μm. The proportions of SFEB (white square) and stromal cell-derived inducing activity (SDIA) (black square) cell culture expressing striatum neuron markers and neurotransmitter-related markers (j). SFEB culture inhibited caudalization activity and produced mature striatum neurons more effectively than SDIA culture. Schema of SDIA and SFEB culture with marker expressions and time schedule of in vivo experiments (k). *P<0.05; **P<0.01.
Selection of Sox1-Positive Neural Progenitors by FACS
We purified the progenitors expressing the neural progenitor marker Sox1 by FACS after 4 days of SFEB culture (Figure 2a). In the GFP-negative group, we confirmed a lack of differentiation activity and the presence of the undifferentiated ES cell markers Oct4 and Nanog-positive cell masses after 5 days of culture in vitro. In contrast, there were no undifferentiated cells in the GFP-positive group (Figure 2b). We have previously confirmed that over 90% of the GFP-positive cells differentiated into Tuj1-positive postmitotic neurons, and half of the GFP-negative cells expressed the Oct4.17

Fluorescence-activated cell sorting (FACS) selection of Sox1-positive embryonic stem (ES) cells and undifferentiated cells in vitro. Flow cytometric profiles of 4-day serum-free suspension culture (SFEB), green fluorescent protein (GFP)-positive and -negative groups, setting of P4 for Sox1-negative cells and P2 for Sox1-positive cells (a). GFP-negative group confirmed undifferentiated ES cell markers Oct4 and Nanog-positive cell mass (b–d), compared with GFP-positive group (e–g). Scale bar: 100 μm.
Prevention of Teratoma Formation In Vivo
We transplanted three different cell groups, GFP-positive (n=20), negative progenitors (n=17), and differentiation medium alone (n=13), into the ischemic putamen at 1 week after focal ischemia. In some cases in the GFP-negative group, we observed that intracranial tumors were bulging out from superficial skin (Figures 3a and b). These enlarged tumors compressed on the brain stem and were fatal owing to cerebral herniation. Kaplan–Meier analysis indicated a significantly higher survival rate in the GFP-positive group than in the GFP-negative group (χ2=3.8861; P=0.0493) (Figure 3j). The tumor bed showed some clusters of undifferentiated ES-like tumor cells that displayed co-positive staining for Oct4 and Nanog, and the proliferation marker Ki67, and this was diagnosed as immature teratoma (Figures 3b–f). In the GFP-positive group, we could hardly detect any aggregation of Ki67-positive cells (GFP-positive group, n=16, 0.028±0.081 mm3; GFP-negative group, n=12, 9.8±16 mm3; P<0.0001) (Figures 3d and g). Furthermore, the scattered Ki67-positive cells in GFP-positive group did not show co-staining with the undifferentiated ES cell markers. Transplanted cells in the GFP-positive group had proliferated within the cerebral ischemia and most of the grafted cells had large cell bodies and extended axons as do mature neurons (Figures 3h and i). Graft survival was not significantly different between GFP-positive and -negative groups (GFP-positive group: 16/20; negative group: 12/17).

In vivo teratoma formation after transplantation. In the green fluorescent protein (GFP)-negative group, relatively large intracerebral tumors, which could cause cerebral herniation, were observed 8 weeks after transplantation (a, b). Histological analysis indicated that these cells (c) were co-stained with the undifferentiated embryonic stem (ES) cell markers, Oct4 and Nanog, and the proliferation marker Ki67, and were diagnosed as immature teratoma (d–f). In the GFP-positive group, clusters of undifferentiated cells were scarcely detected (g). The β-galactocerebroside (β-gal)-positive grafted cells within the cerebral ischemia had large cell bodies and extended axons (h, i). Kaplan–Meier analysis revealed a significantly higher survival rate in the GFP-positive group than in the GFP-negative group (j). Scale bar: b, d, g and h, 1 mm; c, 100 μm; e, f and i, 10 μm. **P<0.01.
Differentiation of Transplanted Sox1-Positive Cells and Neurological Function
At 2 months after transplantation, GFP-positive neural progenitors displayed the mature neuronal markers, NeuN and Tuj1 (Figures 4a and b). Various basal ganglion neuronal markers, DARRP32, parvalbumin, and calbindin, and neurotransmitter-related markers, GAD and serotonin, were also detected (Figures 4c–g, Table 1). In addition, we confirmed the restructuring of neuronal networks using synaptophysin and FG (Figures 4h and i, Table 1 and Supplementary Figure). Although neurological disorders remained unchanged until 5 weeks after transplantation, the GFP-positive group exhibited more prominent recovery in the corner test (GFP-positive group, n=12; GFP-negative group, n=12; P=0.0077, 0.015, 0.0069; days 42, 49, 56, respectively), which estimates sensorimotor function, although the body swing test did not detect any difference among the groups (Figures 5a and b).

Expression of differentiated striatum neuron markers and neurotransmitter-related markers. Immunohistochemical analysis indicates that graft cells (β-galactocerebroside (β-gal)-positive, green) differentiated into postmitotic neurons (Neun and Tuj1 positive, red) (a and b). Colocalization of graft cells and mature striatum neuron markers (DARRP32, parvalbumin, calbindin, and glutamate decarboxylase (GAD) positive, red) shows that graft-derived cells survived and differentiated into mature striatum neurons (c–f). Graft cells were also stained with synaptophysin and Fluorogold (FG) (g and h). Yellow arrows showed colocalized cells. Scale bar: 10 μm.

Function of embryonic stem (ES) cell-derived neural progenitors in a transient focal ischemic model. Although the body swing test showed no difference, in the corner test, functional recovery was detected in the green fluorescent protein (GFP)-positive group compared with the GFP-negative group and medium group (a and b). *P<0.05; **P<0.01.
DISCUSSION
In this study, we demonstrated network formation between the striatum and substantia nigra and functional recovery in the corner test by transplantation of striatum neurons specially induced from mouse ES cells using SFEB culture. We also confirmed that neural progenitors expressing Sox1 did not induce tumorigenesis in the ischemic brain at least for 8 weeks.
Although various methods to improve survival and function of the graft were tested recently, the appropriate protocols for transplantation and their mechanisms have not yet been established. Zhang et al24 reported that human ES cells transplanted into the peri-infarct area migrated to the ischemic core and activated endogenous neurogenesis.
Some transplantation protocols apply to the early phase of ischemia,8 or through the use of hematopoietic progenitor cells can depend on the trophic or growth factor secretion to reduce ischemic injury.25
Our original purpose is to achieve functional recovery by cell replacement at the chronic stage. Thus, we have to improve our method of transplantation by overcoming various challenges, including proper cell induction, teratoma formation, and long-term functional recovery. Furthermore, before clinical trials can begin, studies using non-human primates are desirable.
ES cells are karyotypically normal, capable of unlimited, undifferentiated proliferation in vitro, and retain the potential to generate all cell types. After modeled cerebral ischemia, and especially MCA occlusion, the striatum and sensorimotor cortex are mainly affected. These areas are part of the telencephalon. In previous reports, we used SDIA method to induce neural progenitor cells.12, 13 This method is efficiently producing significant numbers of midbrain dopaminergic neurons.19 Retinoic acid treatment also caudalizes neural progenitors in embryoid bodies.26 Watanabe et al14 demonstrated directed differentiation of telencephalic progenitors from mouse ES cells using SFEB culture with Wnt and Nodal antagonists. In our study, we applied this method to induce striatum neurons through telencephalic neural progenitors from ES cells. Thus as indicated in the results, SFEB could decrease levels of Pax2-positive neurons and induce more DARRP32- and GAD65-positive neurons both in vitro and in vivo. These latter markers are expressed in GABAergic medium spiny neurons, mainly striatum neurons; we transplanted cells into the lateral striatum. In this sense, SFEB culture is a suitable method for transplantation in treatment of MCA occlusion.
Another concern of transplantation using ES cell-derived cells is tumorigenesis. Undifferentiated cells are well known as being the cause of teratoma formation, and to discriminate at which stage the cells lose pluripotency is crucially important. Bieberich et al27 found that ceramide-induced apoptosis eliminates pluripotent cells expressing Oct4. In this study, we employed the neural progenitor marker Sox1 as the earliest-known specific marker of neuroectoderm in the mouse embryo; the purification of neuroepithelial cells by FACS using this marker has been reported previously.18 We have previously used Sox1 as a marker and analyzed tumorigenesis after transplantation into healthy mice.17 In this study, we attempted to use FACS sorting in a model of stroke. FACS sorted graft did not show teratoma formation with few Ki67-positive cells, which were not stained for undifferentiated ES cell markers. We regard these scattered proliferating cells as residual Sox1-positive neural progenitors, and these cells would not form tumors as clusters of ES-like cells. However, we could not conclude that these cells would prevent tumor formation after 2 months transplantation. Long-term observation would be necessary to determine the safety of Sox1-positive cells.
Furthermore, we examined the function of transplanted cells by immunohistochemical analysis for synaptophysin and FG infusion. This is the same technique as used in our previous xenograft transplantation.13 Synaptophysin is a synaptic vesicle glycoprotein in synaptic transmission and FG is a retrograde tracer. Synaptophysin- or FG-positive transplanted cells could indicate that these cells form neuronal network, even with infused substantia nigra. According to the data showed in this study, we cannot completely exclude the possibility of FG spillage during injection procedure. It is necessary to perform further experiment to confirm the connection between the transplanted cells and substantia nigra. Long-term impairments after MCA occlusion are difficult to assess because of spontaneous recovery, especially in mouse models. Zhang et al22 reported a correlation between the infarct volume and the sensorimotor impairments measured by the corner test 3 months after mouse MCA occlusion. We confirmed functional recovery in corner test in GFP-positive group, compared with GFP-negative group. There is a possibility that the repaired neural circuits detected by immunohistochemical analysis contributed to the functional benefit. In contrast, Body swing test focusing on motor weakness as a striatal test did not show the difference among groups. The corner test can detect long-term sensorimotor dysfunction, which involves both stimulation of the vibrissae (sensory/neglect) and rearing (motor response). Besides a test sensitivity, our cell therapy may mainly connect the recovery of sensory disturbance.
Conclusion
We demonstrated network formation between the striatum and substantia nigra and functional recovery in the corner test by transplantation of striatum neurons specially induced from mouse ES cells using SFEB culture. We also confirmed that neural progenitors expressing Sox1 did not induce tumorigenesis in the ischemic brain at least for 8 weeks.
References
Bjorklund A, Lindvall O . Cell replacement therapies for central nervous system disorders. Nat Neurosci 2000;3:537–544.
Savitz SI, Dinsmore JH, Wechsler LR, et al. Cell therapy for stroke. NeuroRx 2004;1:406–414.
Lindvall O, Kokaia Z . Stem cells for the treatment of neurological disorders. Nature 2006;441:1094–1096.
Arvidsson A, Collin T, Kirik D, et al. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med 2002;8:963–970.
Ikeda R, Kurokawa MS, Chiba S, et al. Transplantation of neural cells derived from retinoic acid-treated cynomolgus monkey embryonic stem cells successfully improved motor function of hemiplegic mice with experimental brain injury. Neurobiol Dis 2005;20:38–48.
Wei L, Cui L, Snider BJ, et al. Transplantation of embryonic stem cells overexpressing Bcl-2 promotes functional recovery after transient cerebral ischemia. Neurobiol Dis 2005;19:183–193.
Kim DY, Park SH, Lee SU, et al. Effect of human embryonic stem cell-derived neuronal precursor cell transplantation into the cerebral infarct model of rat with exercise. Neurosci Res 2007;58:164–175.
Takahashi K, Yasuhara T, Shingo T, et al. Embryonic neural stem cells transplanted in middle cerebral artery occlusion model of rats demonstrated potent therapeutic effects, compared to adult neural stem cells. Brain Res 2008;1234:172–182.
Hicks AU, Lappalainen RS, Narkilahti S, et al. Transplantation of human embryonic stem cell-derived neural precursor cells and enriched environment after cortical stroke in rats: cell survival and functional recovery. Eur J Neurosci 2009;29:562–574.
Smith AG . Embryo-derived stem cells: of mice and men. Annu Rev Cell Dev Biol 2001;17:435–462.
Buhnemann C, Scholz A, Bernreuther C, et al. Neuronal differentiation of transplanted embryonic stem cell-derived precursors in stroke lesions of adult rats. Brain 2006;129:3238–3248.
Takagi Y, Nishimura M, Morizane A, et al. Survival and differentiation of neural progenitor cells derived from embryonic stem cells and transplanted into ischemic brain. J Neurosurg 2005;103:304–310.
Hayashi J, Takagi Y, Fukuda H, et al. Primate embryonic stem cell-derived neuronal progenitors transplanted into ischemic brain. J Cereb Blood Flow Metab 2006;26:906–914.
Watanabe K, Kamiya D, Nishiyama A, et al. Directed differentiation of telencephalic precursors from embryonic stem cells. Nat Neurosci 2005;8:288–296.
Erdo F, Buhrle C, Blunk J, et al. Host-dependent tumorigenesis of embryonic stem cell transplantation in experimental stroke. J Cereb Blood Flow Metab 2003;23:780–785.
Harkany T, Andang M, Kingma HJ, et al. Region-specific generation of functional neurons from naive embryonic stem cells in adult brain. J Neurochem 2004;88:1229–1239.
Fukuda H, Takahashi J, Watanabe K, et al. Fluorescence-activated cell sorting-based purification of embryonic stem cell-derived neural precursors averts tumor formation after transplantation. Stem Cells 2006;24:763–771.
Aubert J, Stavridis MP, Tweedie S, et al. Screening for mammalian neural genes via fluorescence-activated cell sorter purification of neural precursors from Sox1-gfp knock-in mice. Proc Natl Acad Sci USA 2003;100:11–11.
Kawasaki H, Mizuseki K, Nishikawa S, et al. Induction of midbrain dopaminergic neurons from ES cells by stromal cell-derived inducing activity. Neuron 2000;28:31–40.
Longa EZ, Weinstein PR, Carlson S, et al. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989;20:84–91.
Borlongan CV, Randall TS, Cahill DW, et al. Asymmetrical motor behavior in rats with unilateral striatal excitotoxic lesions as revealed by the elevated body swing test. Brain Res 1995;676:231–234.
Zhang L, Schallert T, Zhang ZG, et al. A test for detecting long-term sensorimotor dysfunction in the mouse after focal cerebral ischemia. J Neurosci Methods 2002;117:207–214.
Krauss S, Johansen T, Korzh V, et al. Expression of the zebrafish paired box gene pax[zf-b] during early neurogenesis. Development 1991;113:1193–1206.
Zhang P, Li J, Liu Y, et al. Transplanted human embryonic neural stem cells survive, migrate, differentiate and increase endogenous nestin expression in adult rat cortical peri-infarction zone. Neuropathology 2009;29:410–421.
Oyamada N, Itoh H, Sone M, et al. Transplantation of vascular cells derived from human embryonic stem cells contributes to vascular regeneration after stroke in mice. J Transl Med 2008;6:54.
Wichterle H, Lieberam I, Porter JA, et al. Directed differentiation of embryonic stem cells into motor neurons. Cell 2002;110:385–397.
Bieberich E, Silva J, Wang G, et al. Selective apoptosis of pluripotent mouse and human stem cells by novel ceramide analogues prevents teratoma formation and enriches for neural precursors in ES cell-derived neural transplants. J Cell Biol 2004;167:723–734.
Acknowledgements
This study was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, and Culture of Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
A new method for generation and isolation of neural progenitors from embryonic stem cells is described. When the cells are transplanted into ischemic mouse putamen after transient middle cerebral artery occlusion, neuronal network formation and functional recovery from ischemic damage is observed.
Supplementary information
Rights and permissions
About this article
Cite this article
Fujimoto, M., Hayashi, H., Takagi, Y. et al. Transplantation of telencephalic neural progenitors induced from embryonic stem cells into subacute phase of focal cerebral ischemia. Lab Invest 92, 522–531 (2012). https://doi.org/10.1038/labinvest.2012.1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2012.1
Keywords
This article is cited by
-
Generation of pluripotent stem cells without the use of genetic material
Laboratory Investigation (2015)

{kind=link}