
Abstract
Mast cell (MC)-deficient c-Kit mutant KitW/W-v mice are protected against experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis, suggesting a detrimental role for MCs in this disease. To further investigate the role of MCs in EAE, we took advantage of a recently characterized model of MC deficiency, KitW-sh/W-sh. Surprisingly, we observed that myelin oligodendrocyte glycoprotein (MOG)35–55-induced chronic EAE was exacerbated in KitW-sh/W-sh compared with Kit+/+ mice. KitW-sh/W-sh mice showed more inflammatory foci in the central nervous system (CNS) and increased T-cell response against myelin. To understand whether the discrepant results obtained in KitW-sh/W-sh and in KitW/W-v mice were because of the different immunization protocols, we induced EAE in these two strains with varying doses of MOG35–55 and adjuvants. Although KitW-sh/W-sh mice exhibited exacerbated EAE under all immunization protocols, KitW/W-v mice were protected from EAE only when immunized with high, but not low, doses of antigen and adjuvants. KitW-sh/W-sh mice reconstituted systemically, but not in the CNS, with bone marrow-derived MCs still developed exacerbated EAE, indicating that protection from disease could be exerted by MCs mainly in the CNS, and/or by other cells possibly dysregulated in KitW-sh/W-sh mice. In summary, these data suggest to reconsider MC contribution to EAE, taking into account the variables of using different experimental models and immunization protocols.
Similar content being viewed by others


Main
MCs are key factors in IgE-associated immediate hypersensitivity reactions, during which they release a wide spectrum of inflammatory mediators in response to IgE and antigen (Ag) stimulation.1 However, recent findings have pointed out that MCs may exert also important effector and/or immunomodulatory functions in other physiopathological conditions as venom detoxification, pathogen clearance, tumor growth, contact hypersensitivity, allograft acceptance and autoimmunity.2 Most of these studies have assessed the in vivo role of MCs by the use of mouse models carrying spontaneous mutations in c-Kit receptor (WBB6F1-KitW/W-v or C57BL/6-KitW-sh/W-sh mice) or c-Kit ligand (WCB6F1-KitlSl/KitlSl-d), which show severe deficiency of MC populations.3, 4
MCs may either enhance or suppress the inflammation associated to different types of autoimmune diseases. MC-deficient KitW/W-v and KitlSl/Sl-d mice are protected from autoantibody-mediated models of rheumatoid arthritis and bullous pemphigoid, inflammatory disorders affecting, respectively, the joint and the skin. Reconstitution of KitW/W-v mice with bone marrow derived, in vitro cultured MCs (BMMCs) restores complete disease susceptibility in both conditions, thus indicating a proinflammatory role of these cells in such diseases.5, 6 Conversely, MCs seem to exert a protective function in experimental autoimmune glomerulonephritis, by limiting clinical and histological glomerular pathology and mortality.7, 8 However, recent work has shown that, unlike KitW/W-v, KitW-sh/W-sh mice develop a full clinical and histological autoimmune arthritis, thus questioning the actual involvement of MCs in this disorder.9
A complex and conflicting scenario emerges from studies on the role of MCs in promoting central nervous system (CNS) autoimmune response occurring in multiple sclerosis (MS) and its animal model, experimental autoimmune encephalomyelitis (EAE).10 Several reports have documented the presence of MCs within the plaques of MS patients, where they are generally clustered around venules and capillaries, in lower numbers in acute lesions than in chronic active plaques.11, 12 Gene microarray and quantitative RT-PCR analyses have revealed that transcripts for MC-related genes (ie, triptase and IgE Fcɛ receptor I) are upregulated in MS plaques13, 14 and MC tryptase is also elevated in the spinal fluid during MS relapses.15 In EAE, numerous studies have reported a correlation between number, distribution and/or activation state of brain MCs and the development and severity of the disease.10 A significant support for a proinflammatory role of MCs in EAE derives from M Brown's group, showing that KitW/W-v mice develop EAE with delayed onset and significantly milder severity as compared with congenic Kit+/+ mice.16 Reconstitution of these mice with BMMCs derived from wild-type (WT) mice but not from mice genetically lacking Fc receptors restores typical disease susceptibility.17 Further studies have demonstrated that MCs may exert their immune functions in this model by influencing autoimmune T-cell responses in periphery, rather than in the parenchyma of CNS, which is not repopulated in KitW/W-v mice after BMMC transplantation.18 In line with these findings, the analysis of draining lymph nodes during acute EAE revealed activated MCs, regulatory T cells and Th17 cells establishing tight spatial interactions, suggesting the occurrence of a MC-mediated inhibition of regulatory T-cell function in support of Th17 immune response.19 In KitW/W-v mice, MCs have also extra-lymphoid roles in EAE, favoring the access of neutrophils to inflamed CNS by secreting TNF in meninges.20
In contrast to these previous works, Bennett et al21 have recently shown that MCs are dispensable for EAE development in both KitW/W-v and KitW-sh/W-sh models. Conversely, Stelekati et al22 have recently published that EAE severity is decreased in KitW-sh/W-sh mice but is restored following MC knockin,22 thus confirming the data obtained by Brown and colleagues in KitW/W-v model.
It is possible that discrepant results obtained on this topic by different groups could in part be related to different protocols of immunization used to elicit EAE, as well as to different animal models of MC deficiency. Indeed, at least in the KitW/W-v model, it has been shown that the doses of adjuvant or peptide in immunization protocols can drastically affect MC involvement in the disease under investigation, as emerged in experimental models of asthma23 and contact hypersensitivity.24
The aim of our study was to unravel the contribution of MCs to EAE in both KitW-sh/W-sh and KitW/W-v strains by using different protocols of immunization, in order to evaluate if disease outcome was affected by different experimental settings. In this study, we report that, under three different immunization conditions, KitW-sh/W-sh mice develop a more severe EAE and an increased immune response against myelin than Kit+/+ animals. However, systemic reconstitution of KitW-sh/W-sh mice with BMMCs fails to restore WT susceptibility, therefore suggesting that in this model peripheral MCs may be redundant for EAE development. Surprisingly, KitW/W-v mice are protected from EAE only when immunized with a ‘strong’ protocol of immunization (ie, with higher doses of Ag and adjuvants, similar to the one used by Secor et al), but not when immunized with a milder one. Thus, MC contribution to EAE pathogenesis needs to be reconsidered, taking into account the variations because of the different immunization conditions and genetic defects.
MATERIALS AND METHODS
Animals
c-Kit mutant C57BL/6-KitW-sh/W-sh mice and their WT controls C57BL/6-Kit+/+, c-Kit mutant WBB6F1-KitW/W-v mice and their congenic WT littermates WBB6F1-Kit+/+, and myelin oligodendrocyte glycoprotein (MOG)35–55-TCR transgenic 2D2 mice were purchased from Jackson Laboratories. Age-matched female, 8- to 12-weeks-old mice, were used in all EAE experiments. Mice were maintained under pathogen-free conditions at the animal facility of Fondazione IRCCS Istituto Neurologico ‘Carlo Besta’ Milano or of Stanford University. All procedures involving animals were approved by the Institute Ethical Committee and performed in accordance to institutional guidelines and national law (DL116/92) and the Division of Comparative Medicine at Stanford University, and carried out according to the Principles of Laboratory Animal Care (European Communities Council Directive 86/609/EEC) and the National Institutes of Health guidelines.
Peptide Synthesis, Protocols of Active and Passive EAE and Granulocyte Depletion
MOG35–55 (MEVGWYRSPFSRVVHLYRNGK) and control peptide (rat P0; DGDFAIVKFTKVLLDYTGHI) were synthesized using a standard 9-fluorenylmethoxycarbonyl chemistry on a 433A automated peptide synthesizer (Applied Biosystems) and purified by HPLC. The purity of each peptide was >95% as assessed by analytical reverse-phase HPLC. EAE was induced with different protocols of immunization. For EAE induction in Figure 1a, MOG35–55 peptide was dissolved in PBS to a concentration of 4 mg/ml and emulsified with an equal volume of IFA supplemented with 8 mg/ml heat-killed Mycobacterium tuberculosis H37Ra (Difco). Mice were injected s.c. in their flanks with 0.1 ml of the peptide emulsion (for a total of 200 μg of MOG35–55/mouse and 400 μg of M. tuberculosis) and, on the same day and at 48 h later, were injected i.v. with 0.2 ml containing 200 ng of Bordetella pertussis Toxin (List Laboratories) dissolved in PBS. Doses of MOG35–55 and adjuvants used in other protocols are specified in figure legends. Passive EAE was induced as described in Yang et al.25 Briefly, splenocytes from 2D2 mice were activated with MOG35–55 (20 μg/ml), IL-2 (Proleukin) and IL-7 (Peprotech) for 2 days, expanded with IL-2 and IL-7 for 5 days, restimulated overnight with coated anti-CD3 (eBioscience), IL-12 (kindly provided by Dr Maurice Gately, Hoffmann LaRoche, Nutley, NJ) and IL-18 (R&D Systems) and i.p. injected (106/mouse). Mice were assessed daily for neurological signs of EAE according to the following five-point scale: (0) healthy; (1) tail weakness or paralysis; (2) paraparesis (incomplete paralysis of one or two hind limbs/plegia of one hind limb); (3) paraplegia extending to the thoracic level; (4) forelimb weakness or paralysis with hind limbs paraparesis or paraplegia; and (5) moribund or dead animal. For granulocyte depletion, mice were treated i.p. with 10 μg anti-Ly6G mAb (clone RB6-8C5) dissolved in saline (200 μl) daily from d0 to d9.

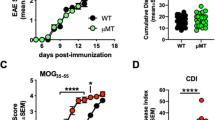
MC-deficient KitW-sh/W-sh mice develop exacerbated EAE associated with increased CNS inflammation and autoimmune response. (a) EAE was induced in Kit+/+ (n=10) and KitW-sh/W-sh (n=17) mice with MOG35–55 peptide as detailed in Materials and Methods and mice were scored daily as described.28 Data represent mean clinical score±s.e.m. (*P<0.05, ***P<0.005, ****P<0.001; Student's t-test, two tailed). These data are from a single experiment representative of three independent replicates, each including 9–17 mice per group. (b) Distribution and phenotype of the lesions in the CNS of Kit+/+ and KitW-sh/W-sh mice obtained at 7 weeks after EAE induction. The upper left images show representative brain sections from cerebellum (left panel) and hippocampal/thalamic region (right panel) from KitW-sh/W-sh mice highlighting severe perivascular infiltrates (asterisk), as shown in detail in the corresponding insets. Conversely, Kit+/+ mice (upper right images) show no signs of active inflammation with few dilated vessels and rare inflammatory cells (insets) both in the cerebellum (left panel) and in the hippocampal/thalamic region (right panel). Similar findings were observed in the spinal cords, with the KitW-sh/W-sh mice showing severe perivascular inflammatory infiltrates (asterisk; left panels) and the Kit+/+ mice being substantially free of inflammatory cells with foci of residual myelin damage (asterisk) with microcystic appearance (inset). (c) Proliferation rate of LNCs (mean c.p.m.±s.e.m., from triplicate wells) and (d) cytokine production of splenocytes (means±s.e.m., from duplicate wells) isolated from Kit+/+ and KitW-sh/W-sh mice (n=4 mice per group) at 10 days after EAE induction and stimulated with MOG35–55, concanavalin A (conA) or medium alone. *P<0.05, **P<0.01, ***P<0.005 (Student's t-test, two tailed). (e) Representative graphs showing percentages of CD4+ IL-17+ and CD4+ IFN-γ+ T cells in draining lymph node obtained from Kit+/+ and KitW-sh/W-sh mice at 10 days post-EAE induction. (f) Draining lymph node isolated from Kit+/+ and KitW-sh/W-sh during acute (d10) and chronic (d48) EAE display a different density of IL-10-expressing mononuclear cells (anti-IL-10, STREPT-ABC complex method; original magnification × 200). (g) Anti-MOG35–55-specific IgG antibodies and IgG antibody isotypes in sera from Kit+/+ (n=10) and KitW-sh/W-sh (n=15) mice obtained at 7 weeks after the induction of EAE. Each mouse was tested individually in duplicate. Bars represent mean titers±s.e.m.
Bone-Marrow-Derived MC Differentiation and Adoptive Transfer into KitW-sh/W-sh Mice
BMMCs were obtained by in vitro differentiation of bone marrow cells taken from mouse femurs as described.26 After 5 weeks, BMMCs were monitored for FcɛRI expression by flow cytometry using FITC-conjugated anti-mouse FcɛRI-α (clone MAR-1, eBioscience) and PE-Cy7-conjugated anti-mouse c-Kit (clone 2B8, eBioscience). Purity was usually >90%. BMMCs were obtained from pooled three to four mice in each experiment. For MC-engraftment studies, BMMCs derived from Kit+/+ mice were injected i.v. into the tail vein (107 cells in 400 μl PBS) into 4-weeks-old female KitW-sh/W-sh mice. EAE experiments were initiated at 6–8 weeks after adoptive transfer of BMMCs.
T-Cell Proliferation Assay and Cytokine Measurement
Draining lymph node cells (LNCs) were isolated from KitW-sh/W-sh and Kit+/+ mice at 10 days after the induction of EAE, and they were cultured in vitro with MOG35–55, Con A (1 μg/ml; positive control), rat P0 (negative control) or medium alone. Cells were cultured in 96-well microtiter plates at a density of 4–5 × 105 cells/well in 200 μl of RPMI 1640 (EuroClone) supplemented with L-glutamine (2 mM), sodium pyruvate (1 mM), nonessential amino acids (0.1 mM), penicillin (100 U/ml), streptomycin (0.1 mg/ml), 2-ME (5 × 105 M), HEPES buffer (0.01 M) and 10% FCS (enriched RPMI 1640). After 72 h of incubation at 37°C with 5% CO2, cultures were pulsed for 18 h with 0.5 μCi of [3H]thymidine per well and proliferation was measured from triplicate cultures on a β-counter (PerkinElmer). Data are expressed as mean c.p.m.±s.e.m. For cytokine analysis, spleen cells were harvested from KitW-sh/W-sh and Kit+/+ mice at 10 days after the induction of EAE and cultured in 24-well plates (3.5 × 106 cells/well) with MOG35–55, Con A, rat P0 or medium alone in the same conditions as described above. Supernatants from in vitro-cultured spleen cells were analyzed by ELISA for IFN-γ, IL-6, IL-10 (anti-mouse OptEIA ELISA Set; BD Pharmingen), and IL-17 (R&D Systems) according to the manufacturer's protocols. Supernatants were collected from cultured cells at different time points: 24 h for IL-6, 48 h for IFN-γ and 96 h for IL-10 and IL-17. Results are shown as mean of duplicates; s.e.m. were always within 10% of the mean.
Flow Cytometry
PE-Cy7-conjugated anti-CD4 (clone L3T4), PE-conjugated anti-CD11b (clone M1/70), FITC-conjugated anti-Gr1 (clone RB6-8C5) were from eBioscience. FITC-conjugated anti-Ly6G (clone 1A8) and APC-conjugated Ly6C (clone 1G7.G10) were from Miltenyi Biotec. Surface staining reactions were performed in PBS supplemented with 2% FCS on ice for 30 min. Intracellular staining with AlexaFluor647-conjugated anti-IL17A (clone eBio17B7) and PE-conjugated anti-IFN-γ (clone XMG1.2) was carried out after 4 h stimulation with Monensin solution (eBioscience), 50 ng/ml PMA and 500 ng/ml ionomycin (Sigma-Aldrich). Staining with PE-conjugated anti-Foxp3 mAb (clone FJK-16S) was performed according to the manufacturer's instructions (eBioscience). Flow cytometry data were acquired on a FACSCalibur (Becton Dickinson) and analyzed with FlowJo software (version 8.8.6; Treestar).
Histopathology and Immunohistochemistry
For histological evaluation of EAE in KitW-sh/W-sh and Kit+/+ mice, five animals per group were killed at 7 weeks after the induction of EAE, and the brain and spinal cord were removed, fixed in 10% formalin and embedded in paraffin. Sections (4 μm) were cut on a microtome and stained with hematoxylin and eosin to study basic histopathological changes. Sections were analyzed in a blinded manner (PLP). The inflammatory changes were expressed as number of perivascular inflammatory infiltrates per square millimeter in the spinal cord27 or as number of inflammatory foci per section, respectively, for the spinal cord and the brain.
Histopathological analysis of MC distribution in steady state and inflamed tissues was carried out on toluidine blue-stained sections from formalin-fixed, paraffin-embedded samples. The number of MCs was estimated by counting MCs out of five high-power microscopic fields ( × 400) showing the highest MC densities and then summing the counts relative to each HPF. For immunohistochemical detection of IL-10+ cells, sections from formalin-fixed, paraffin-embedded lymph nodes underwent immunostaining with the streptavidin biotin peroxidase complex method using a specific primary anti-mouse IL-10 antibody (clone JES5-2A5, eBioscience) and aminoethylcarbazole as chromogen. All the immunostained sections were counterstained with hematoxylin. Slides were analyzed under a Leica DM2000 optical microscope, and microphotographs were collected using a Leica DFC320 digital camera.
Measurement of Serum Antibody Response
Blood was collected from the tail of KitW-sh/W-sh and Kit+/+ mice before and at 7 weeks after the induction of EAE. MOG35–55-specific IgG, IgG1, IgG2a, IgG2b and IgG3 Abs were measured by ELISA as described elsewhere.28 Briefly, 96-well microtiter plates (Immunol; Thermo Labsystems) were coated overnight at 4°C with 0.1 ml of MOG35–55 diluted in 0.1 M NaHCO3 buffer (pH 9.5) at a concentration of 0.010 mg/ml. The plates were blocked with PBS/10% FCS (blocking buffer) for 2 h. Samples were diluted in blocking buffer at 1/100, and Ab binding was tested by the addition of peroxidase-conjugated monoclonal goat anti-mouse IgG, IgG1, IgG2a, IgG2b and IgG3 (Southern Biotechnology Associates), each at a 1/5000 dilution in blocking buffer. Enzyme substrate was added and plates were read at 450 nm on a microplate reader.
Statistical Analysis
Student's t-test, two tailed, was used to compare the results between the two groups. In all tests, P<0.05 was considered statistically significant.
RESULTS
MC-Deficient KitW-sh/W-sh Mice Develop Exacerbated EAE
We first evaluated the involvement of MCs in EAE by using the more recently characterized model of MC deficiency, KitW-sh/W-sh mice, which seems to bear milder c-Kit-related phenotypic abnormalities than KitW/W-v strain.29 Indeed, KitW-sh/W-sh mice are not anemic, neutropenic or sterile as KitW/W-v animals, although they are affected by some hematopoietic alterations such as splenomegaly with expanded myeloid populations, and increased number of circulating neutrophils and platelets.30 Chronic progressive EAE was induced in MC-deficient KitW-sh/W-sh and Kit+/+ mice by immunization with MOG35–55. This protocol induces in this strain a clinical course classically characterized by the acute onset of neurological symptoms reaching a peak about at 2 weeks after immunization, followed by a short and generally incomplete recovery phase after which symptoms of intermediate gravity are chronically maintained.28, 31 Surprisingly, KitW-sh/W-sh mice developed exacerbated EAE compared with Kit+/+ mice (Figure 1a, Table 1), characterized by an anticipated onset and a more severe progression. The clinical severity of EAE was significantly worsened in KitW-sh/W-sh mice during both recovery and chronic phases of disease (mean disease score was 1.8±0.2 in Kit+/+ vs 2.8±0.3 in KitW-sh/W-sh mice (P<0.05) at day 20 (recovery phase), and 1.8±0.2 in Kit+/+ vs 2.6±0.3 in KitW-sh/W-sh mice (p<0.05) at day 43 (chronic phase)). The clinical data were corroborated by a histopathological analysis of CNS inflammation in the chronic phase that revealed more inflammatory foci in the CNS of KitW-sh/W-sh compared with Kit+/+ mice (2.17±1.15 vs 0.75±0.79/mm2, respectively, P<0.05, in the spinal cord; 2.08±1.49 vs 0.83±0.76/section, respectively, P>0.05, in the brain; Figure 1b).
MCs can limit the magnitude of some adaptive immune responses by establishing functional interactions with regulatory T cells32 or by secreting IL-10.33 Given the increased EAE severity shown by KitW-sh/W-sh mice, we investigated whether the peripheral immune response against myelin was affected by MC deficiency. For this purpose, we isolated draining lymph nodes and spleens from mice with EAE at 10 days after immunization. LNCs from KitW-sh/W-sh mice showed a significantly increased proliferation in response to MOG35–55 (Figure 1c) and Ag-stimulated splenocytes from these mice produced significantly more IL-17 and IL-6, but less IL-10, than Kit+/+ animals (Figure 1d). There were no significant differences in the production of IFN-γ (Figure 1d). Percentages of IL-17+ CD4 T cells (0.9±0.4 in Kit+/+ vs 2.2±0.2 in KitW-sh/W-sh mice, P<0.05) and IFN-γ+ CD4 T cells (1.2±0.2 in Kit+/+ vs 2.5±0.6 in KitW-sh/W-sh mice, P>0.05) were also increased in lymph nodes from KitW-sh/W-sh mice compared with Kit+/+ animals (Figure 1e). Immunohistochemical analysis of lymph node sections revealed few scattered IL-10+ cells in KitW-sh/W-sh and control Kit+/+ samples from the acute phase. In these samples, a slight difference was observed in the amount of IL-10+ mononucleated cells between KitW-sh/W-sh and Kit+/+ mice. Such a difference was more evident in samples from the chronic phase, in which the overall amount of IL-10+ cells was higher (Figure 1f). In line with these results, intracellular staining of IL-10 showed a slightly decreased IL-10 content in CD4 T cells in KitW-sh/W-sh mice in both the acute and chronic phases (Supplementary Figure S1).
MCs have been described to enhance IgE and IgG1 production by B cells34, 35 and to secrete cytokines affecting B-cell responses such as IL-4, IL-5 and IL-13.1 Analysis of Ab responses in sera from immunized KitW-sh/W-sh and Kit+/+ mice (Figure 1g) revealed a slight but not significant decrease in titers of IgG antibodies specific for MOG35–55 in MC-deficient mice (mean OD, 0.190±0.074 in Kit+/+ vs 0.079±0.047 in KitW-sh/W-sh mice; P=0.194). Analysis of the IgG isotypes showed decreased titers of Ag-specific IgG2b Abs in KitW-sh/W-sh compared with Kit+/+ mice (mean OD, 0.890±0.477 in Kit+/+ vs 0.177±0.124 in KitW-sh/W-sh mice, P=0.178) but not of the other IgG isotypes.
We19, 26 and others32, 36, 37 have reported that MCs can arrange several kinds of interaction with Treg. We tested whether KitW-sh/W-sh mice harbored some alteration in the Treg compartment. We found a significantly lower Treg frequency in spleens, but not in draining lymph nodes, of KitW-sh/W-sh compared with Kit+/+ mice, at both d10 and d42 after immunization (Supplementary Figure S2). However, the fraction of Treg among CNS-infiltrating CD4 T cells at d10 post immunization did not differ between KitW-sh/W-sh and controls (Supplementary Figure S2C). A decrease in Treg frequency was also observed in lymph nodes of naive KitW-sh/W-sh compared with Kit+/+ mice (Supplementary Figure S3).
We evaluated the amount and distribution of MCs by histopathological evaluation of toluidine-blue-stained sections. In both naive and immunized Kit+/+ mice only few MCs were detected in the CNS, mostly associated with the meninges. MCs infiltrating the brain or spinal cord parenchyma were only exceptionally observed in immunized Kit+/+ mice. The amount and distribution of MCs in draining lymph nodes were affected by immunization, as lymph nodes from naive mice showed few MCs mainly lodged inside the sinuses or within the medullary area (not shown), whereas those from immunized mice showed higher numbers of MCs either scattered or clustered within the T-cell-rich perifollicular areas (Figure 2). In naïve and immunized KitW-sh/W-sh mice, no MCs were detected in the CNS parenchyma. Draining lymph nodes from the same mice were characterized by a hypertrophic medulla with dilated sinuses and by the absence of MCs (Figure 2).

Mast cell distribution in brain, spinal cord and draining lymph node (DLN) of Kit+/+ and KitW-sh/W-sh EAE mice. In immunized Kit+/+ mice few MCs populate the CNS, mostly localized within the meninges. In DLNs from the same mice, small clusters of MCs can be observed in the perifollicular areas of the cortex. In KitW-sh/W-sh mice, no MCs can be detected in either the CNS or DLNs. (Toluidine blue; original magnification × 100, inset × 400.)
KitW-sh/W-sh Mice Develop Severe EAE Regardless the Immunization Protocol
Clinical and immunological findings obtained by us in the KitW-sh/W-sh model were in contrast with the work by Secor et al16 showing that MC-deficient KitW/W-v mice develop a milder EAE associated with a reduced proinflammatory autoimmune response.38 However, in these studies, a ‘strong’ protocol of immunization (ie, high doses of MOG35–55, heat-killed Mycobacterium tuberculosis in CFA and Bordetella pertussis toxin (PTX)) was used to induce EAE. To evaluate whether the discrepant outcome obtained by us in the KitW-sh/W-sh strain could depend on the specific conditions of immunization, we induced EAE in KitW-sh/W-sh and Kit+/+ mice by two additional protocols of immunization stronger than the one used in Figure 1, containing increased doses of PTX or, similarly to Secor et al, higher doses of MOG35–55, M. tuberculosis and PTX. We observed that KitW-sh/W-sh mice developed a more severe EAE than controls (Figure 3a and b) also under these conditions of immunization. Interestingly, immunization with the strongest protocol (Figure 3b) resulted in a more significant clinical difference between KitW-sh/W-sh and Kit+/+ mice. Analysis of the peripheral immune response against MOG35–55 during the acute phase of disease induced with both protocols of immunization confirmed an increased proinflammatory profile of immune cells from KitW-sh/W-sh compared with those from Kit+/+ mice (data not shown).

KitW-sh/W-sh mice develop exacerbated EAE under different conditions of immunization. EAE was induced by two additional protocols of immunization bearing increased doses of sole PTX or of both adjuvants and myelin peptide. (a) Kit+/+ (n=10) and KitW-sh/W-sh (n=9) were immunized by s.c. injection of 200 μg of MOG35–55 emulsified in complete Freund's adjuvant containing 400 μg of M. tuberculosis (on day 0) and i.v. injection of 400 ng of PTX (on days 0 and +2). (b) Kit+/+ (n=9) and KitW-sh/W-sh (n=10) were s.c. injected with 300 μg of MOG35–55 emulsified in complete Freund's adjuvant containing 500 μg of M. tuberculosis (on days 0 and +7) and i.v. injected with 400 ng of PTX (on days 0 and +2). Mice were scored daily. Data represent mean clinical score±s.e.m. from a representative experiment out of two independent replicates, each including 4–10 mice per group, carried out for each protocol of immunization (*P<0.05, **P<0.01, ***P<0.005, ****P<0.001; Student's t-test, two tailed).
MCs Promote or Inhibit EAE in KitW/W-v Mice Depending on the Conditions of Immunization
Given the surprising observation of exacerbated EAE in absence of MCs in the KitW-sh/W-sh mouse model, we decided to go back to the KitW/W-v mouse model and to study the disease in these mice by using different protocols of immunization as we did in the KitW-sh/W-sh mice. We induced EAE with our standard protocol of immunization or with a ‘stronger’ one similar to that used by Secor et al.16 Surprisingly, with lower doses of Ag and adjuvants, KitW/W-v mice exhibited a more severe disease course than controls, resembling the phenotype observed in the KitW-sh/W-sh strain (Figure 4a). Conversely, when we applied a ‘stronger’ protocol of immunization, KitW/W-v mice developed a significantly milder EAE compared with Kit+/+ mice, confirming a detrimental role for MCs in EAE under these specific experimental conditions (Figure 4b).

KitW/W-v susceptibility to EAE depends on the protocols of immunization. (a) Kit+/+ (n=15) and KitW/W-v (n=15) were immunized with 100 μg of MOG35–55 emulsified in complete Freund's adjuvant containing 200 μg of M. tuberculosis (s.c. on day 0) and 200 ng of PTX (i.v. on days 0 and +2). (b) Kit+/+ (n=10) and KitW/W-v (n=10) were immunized as described in Figure 3b. Mice were scored daily. Data represent mean clinical score±s.e.m. from a single experiment performed for each protocol of immunization (*P<0.05, **P<0.01; Student's t-test, two tailed).
Systemic Reconstitution of KitW-sh/W-sh Mice with BMMCs Fails to Revert EAE Phenotype
To test whether the different EAE severity of Kit+/+ and KitW-sh/W-sh mice was truly due to the absence of MCs, rather than to other c-Kit-related alterations,30 KitW-sh/W-sh mice were engrafted with BMMCs differentiated from Kit+/+ donors. Intravenous route of transplantation allowed repopulation of peripheral lymphoid organs, although, as previously observed,29, 39 MC reconstitution did not restore the physiological MC distribution observed in WT mice (Figure 5a). Also, BMMC i.v. transplantation failed to repopulate the CNS (Figure 5a), as already reported in BMMCs-reconstituted KitW/W-v mice.18 Of note, we observed that Treg deficiency was corrected in lymph nodes of BMMCs-reconstituted naive KitW-sh/W-sh mice (Supplementary Figure S3). As shown in Figure 5c, BMMCs-transplanted KitW-sh/W-sh mice developed EAE similarly to MC-deficient KitW-sh/W-sh mice. These data suggest that in this model the restoration of a peripheral pool of MCs is not sufficient to recapitulate WT disease course, at least during the priming phase.

Mast cell reconstitution fails to attenuate EAE clinical severity in KitW-sh/W-sh mice. (a, b) BMMCs were engrafted i.v. (107 cells/mouse) in KitW-sh/W-sh mice, and MC distribution in multiple organs was analyzed at 6–8 weeks later by toluidine blue staining. (a) Intravenously-injected MCs repopulated tissues of KitW-sh/W-sh mice and preferentially accumulated in the spleen, liver and lung. The number of MCs was estimated by counting MCs in five high-power microscopic fields ( × 400) showing the highest MC densities and then summing the counts relative to each high-power field. Data represent mean cell counts in each organ±s.e.m. (*P<0.05, ***P<0.005; Student's t-test, two tailed, calculated between Kit+/+ and BMMCs-engrafted KitW-sh/W-sh mice). (b) Inguinal LN (IngLN) from reconstituted KitW-sh/W-sh mice showed a comparable amount of MCs with LN from WT mice, though the distribution of MCs in repopulated LNs was mainly intrasinusoidal (toluidine blue; original magnification × 100, inset × 400). (c) EAE was induced in Kit+/+, KitW-sh/W-sh and BMMCs-transplanted KitW-sh/W-sh mice with MOG35–55 peptide as described in Materials and Methods. BMMCs were injected i.v. at 6–8 weeks before immunization. These data are from a single experiment representative of two independent replicates, each including 4–10 mice per group. (d) Passive EAE was induced in Kit+/+ (n=7) and KitW-sh/W-sh (n=10) mice by i.p. injection of in vitro primed MOG35–55-specific TCR-transgenic 2D2 T cells. Mice were scored daily. Data represent mean clinical score±s.e.m. from a representative experiment out of two independent replicates, each including 7–10 mice per group (*P<0.05, **P<0.01, ***P<0.005; Student's t-test, two tailed).
Passive EAE is Exacerbated in KitW-sh/W-sh Mice
The above data indicated that c-Kit mutation affects the development of autoimmune manifestations mainly outside the peripheral lymphoid organs, suggesting that an altered encephalitogenic T-cell priming was unlikely responsible for the exacerbated disease course observed in KitW-sh/W-sh mice. To confirm this hypothesis, passive EAE was induced in KitW-sh/W-sh and Kit+/+ mice by transferring in vitro primed MOG35–55-specific TCR-transgenic 2D2 T cells.25 This approach allowed uncoupling priming and effector phase of the disease and isolating the role played by c-Kit mutation in inflammation in the CNS. We found that, also in this setting, KitW-sh/W-sh mice developed clinical signs of disease with anticipated onset and exacerbated severity compared with Kit+/+ mice (Figure 5d). This result suggested that the major contribution of the KitW-sh/W-sh mutation to EAE pathology occurred downstream T-cell priming and concerned probably the effector phase of the disease mounting in the CNS.
Alterations in the Granulocyte Compartment do not Influence EAE in KitW-sh/W-sh Mice
KitW-sh/W-sh mutation may affect the homeostasis of granulocyte populations expressing c-Kit.29, 30 As granulocytes participate as effector cells in autoimmune inflammation in the CNS during EAE,40, 41 we speculated that enhanced granulocytosis might contribute to disease exacerbation in KitW-sh/W-sh mice. We first characterized the percentage of Gr1hi CD11bhi and Gr1int CD11bint myeloid populations in the lymphoid organs of naive Kit+/+ and in KitW-sh/W-sh animals. We observed a higher percentage of both subsets in the spleens of KitW-sh/W-sh mice compared with controls, according to previous findings,30 whereas only the Gr1hi CD11bhi population was expanded in their lymph nodes (Supplementary Figure S4A). The anti-Gr1 mAb used for staining (RB6-8C5 clone) recognized both Ly6G+ and Ly6C+ subsets,42 representing granulocytic and monocytic cells, respectively. The analysis of these two populations showed that both Ly6G+ and Ly6C+ cells were expanded in the spleens of KitW-sh/W-sh mice, but only the Ly6G+ CD11b+ subset was amplified in lymph nodes (Supplementary Figure S4B). The Gr1hi CD11bhi population expressed c-Kit at higher level than the Gr1int CD11bint counterpart in Kit+/+ mice; moreover, c-Kit expression in the Gr1hi CD11bhi compartment was significantly decreased in KitW-sh/W-sh mice, whereas remaining unaltered within the Gr1int CD11bint counterpart (Supplementary Figure S4C). These pieces of evidence suggested that Gr1hi CD11bhi cells (likely encompassing the Ly6G+ granulocytes) were more strictly dependent on c-Kit signal for their homeostasis than the Gr1int CD11bint cells (including the Ly6C+ monocytes). To explore the effects of KitW-sh/W-sh mutation in the expansion of myeloid cells upon EAE induction, we looked at the frequency of CD11b+, Ly6G+ and Ly6C+ cells in lymphoid organs and CNS on immunization. The percentage of total CD11b+ cells was higher in draining lymph nodes of KitW-sh/W-sh mice compared with controls at d10 after immunization. A faster accumulation of the Ly6G+, rather than the Ly6C+, fraction mainly accounted for this difference between the two strains (Figure 6a and b). At d10, the leukocytes infiltrating the CNS of KitW-sh/W-sh mice contained a higher fraction of Ly6G+ CD11b+ cells, suggesting that an enhanced granulocyte-mediated inflammation might correspond to a peripheral granulocyte expansion (Figure 6c and d). To identify the contribution of granulocyte expansion to the increased disease severity expressed by KitW-sh/W-sh mice, active EAE was induced in MC-deficient and WT mice under a granulocyte-depleting regimen. To this aim, mice were treated i.p., daily from d0 to d9, with the anti-Gr1 RB6-8C5 clone, which specifically depleted Ly6G+ while sparing Ly6C+ cells (Figure 6f), according to published results.43 In line with previous findings,41 granulocyte elimination importantly delayed the onset and attenuated the severity of EAE symptoms in Kit+/+ mice (Figure 6e). However, although Ly6G+-depleted KitW-sh/W-sh mice developed a milder disease than Kit+/+ untreated controls, these mice still developed a more severe disease than Ly6G+-depleted Kit+/+ controls, suggesting a granulocyte-independent mechanism behind enhanced autoimmune inflammation in c-Kit mutated mice (Figure 6e). In support of this conclusion, we observed that in BMMC-i.v. reconstituted KitW-sh/W-sh mice, in which the normal disease course was not rescued (Figure 5c), alterations of granulocyte homeostasis were completely recovered in both the Gr1hi and Gr1int compartments (Supplementary Figure S5). We noticed decreased T-cell proliferation and inflammatory cytokine production in both Ly6G+-depleted KitW-sh/W-sh and Kit+/+ compared with untreated Kit+/+ mice. This suggests that the increased disease severity observed in granulocyte-depleted KitW-sh/W-sh compared with granulocyte-depleted Kit+/+ mice was not due to differences in T-cell peripheral activation (Figure 6g). Taken together, these data suggest that KitW-sh/W-sh mice developed exacerbated EAE independently from T-cell priming, peripheral MC presence or granulocytic effector cell alterations.

The severe abnormalities in granulocyte compartment of KitW-sh/W-sh mice are not responsible for increased EAE severity. (a–d) The percentage of total CD11b+ and the fraction of Ly6G+/Ly6C+ cells among the CD11b+ compartment was evaluated by flow cytometry in inguinal lymph nodes (a, b) or CNS (c, d) from Kit+/+ and KitW-sh/W-sh mice at days 0, 4 or 10 after immunization. Data in a and c represent mean percentage±s.e.m. (*P<0.05, **P<0.01, ***P<0.005; Student's t-test, two tailed). Representative contour plots of Ly6C vs Ly6G in gated CD11b+ cells are depicted in b and d. (e) Kit+/+ (n=14) and KitW-sh/W-sh (n=6) mice were immunized with MOG35–55 peptide. Kit+/+ (n=7) and KitW-sh/W-sh (n=6) were treated with anti-Ly6G mAb as described in Materials and Methods. The second cohort of Kit+/+ mice (n=7) was not treated and served as an internal control of EAE. Mice were scored daily. Data are from a single experiment representative of two independent replicates, each including six to seven mice per group. Data represent mean clinical score±s.e.m. (*P<0.05, **P<0.01, ***P<0.005; Student's t-test, two tailed, calculated between a-Ly6G-treated Kit+/+ and a-Ly6G-treated KitW-sh/W-sh mice). (f) Representative contour plots showing depletion of Ly6G+ but not Ly6C+ cells in peripheral blood at 8 days after a-Ly6G treatment. (g) Proliferation rate of LNCs (mean c.p.m.±s.e.m., from triplicate wells) and cytokine production of splenocytes (means±s.e.m., from duplicate wells) isolated from untreated Kit+/+ mice and from anti-Gr1-treated Kit+/+ and KitW-sh/W-sh mice (n=3–7 mice per group) at 10 days after EAE induction and stimulated with MOG35–55 10 μg/ml or medium alone. **P<0.01, ***P<0.005 (Student's t-test, two tailed, calculated in comparison with untreated Kit+/+ mice).
Beside MCs, other cells of the immune system may express c-Kit at variable levels and under different circumstances,44 such as dendritic cells (DCs).45 C-Kit pharmacological inhibition fosters a productive cross-talk between DCs and natural killer (NK) cells 46 and promotes the differentiation of a DC subset endowed with cytotoxic ability and IFN-γ competence.47 However, as previously reported,29 we did not observe any difference between naive KitW-sh/W-sh and Kit+/+ mice in NK or DC frequency in the spleen (Supplementary Figure S6).
DISCUSSION
Broad evidence suggests that MCs may have multiple and even divergent roles in autoimmune and inflammatory diseases. The vast majority of these observations come from rodent MC-deficient models harboring different mutations in the c-Kit gene. In this study, we showed that the KitW/W-v animals were differentially susceptible to EAE induction depending on the strength of the immunization protocol. Conversely, KitW-sh/W-sh mice developed anticipated and exacerbated EAE, either passively or actively induced, under all the tested conditions of immunization. Such behavior could not be attributed to an immunoregulatory role of MCs in T-cell priming occurring in lymphoid organs, as indicated by the failure of recovering the normal disease course by reconstituting the peripheral MC pool or by passively inducing EAE bypassing T-cell priming. We confirmed some defects in the granulocyte compartment in KitW-sh/W-sh mice that might participate to inflammation in the CNS, although granulocyte elimination could not abrogate the difference in disease outcome between c-Kit mutant and WT mice.
Most of the information about the role of MCs in EAE came from the Brown's group that showed decreased EAE onset and severity in KitW/W-v mutant mice.16, 38 In their system, MC systemic reconstitution restored disease severity by recovering an efficient encephalitogenic T-cell response in peripheral lymphoid organs18 and by restoring neutrophil penetration at the blood–brain barrier.20 Our results reconciled, in part, the apparently contrasting findings in the two MC-deficient strains. Indeed, we could reproduce Brown's data in the KitW/W-v model when a strong immunization protocol was applied, but under milder conditions, which are our standard procedure to induce active EAE,48 KitW/W-v mice developed an exacerbated rather than an attenuated disease, replicating our results in the KitW-sh/W-sh model. However, our results are in contrast with a recent report from the same group, showing decreased EAE symptoms in KitW/W-v mice compared with Kit+/+ mice also on the application of a relatively milder immunization protocol (100 μg of MOG35–55 and 250 ng of PTX).20 Thus, the discrepancies between our results and those reported by the Brown's group remain to be fully understood. However, it is possible that the opposite results observed in KitW/W-v under different immunization protocols are related to a differential impact of the same mutation on distinct pathological mechanisms depending on the quality/quantity of the immune stimulation and on experimental conditions. Furthermore, different mutations in the same gene/locus can lead to contrasting outcomes under the same conditions of stimulation. Indeed, high doses of MOG35–55, CFA and PTX produced a milder outcome in KitW/W-v mice, but a more severe disease in KitW-sh/W-sh mice than in Kit+/+ controls. The reasons for this discrepancy may rely on differences between strains regarding non-MC-related abnormalities. For instance, it has been shown that KitW/W-v, but not KitW-sh/W-sh, mice are resistant to antibody-induced arthritis because the former is neutropenic, whereas the latter shows splenic myeloid hyperplasia, both at basal level and on immunization.9, 30 Depletion of Gr1+ cells showed a prominence of granulocytes, rather than MCs, in the pathogenetic mechanisms driving arthritis in this model.9 The divergent susceptibility of the two strains to EAE development may even reside in non-immune-related processes. The observation that the KitW-sh/W-sh mutation determines the disruption of the Corin gene30 suggests that the KitW-sh/W-sh model may recapitulate the defects observed in Corin−/− mice. Indeed, KitW-sh/W-sh mice show cardiomegaly30 and may also display spontaneous hypertension, a condition associated to Corin ablation49 that may favor EAE development, as indicated by the recent observation that anti-hypertensive drugs suppress autoimmune response in EAE.50 The KitW-sh/W-sh mutation may also produce dysregulations in a series of genes included in the inverted region, whose effects in immunity are largely unknown.30 Finally, the two c-Kit-mutated strains differ in their genetic background, and the WBB6F1-Kit+/+ is characterized by higher number of peritoneal MCs and neutrophils than the C57BL/6-Kit+/+.51 In summary, our findings underscore the need for clear and doubtless identification of the role of MCs in a certain process by taking advantage of different c-Kit mutants. These variables (the strain of MC-deficient mice, the kind of mutation in c-Kit gene and the experimental model) are well exemplified by the work of Piliponsky et al52 in mouse models of sepsis. This study demonstrates that MCs can promote survival in cecal ligation and puncture (CLP) of moderate severity in WBB6F1-KitW/W-v, C57BL/6-KitW-sh/W-sh and WBB6F1-KitW-sh/W-sh strains, but they exert no effect in WBB6F1-KitW/W-v and WBB6F1-KitW-sh/W-sh affected by severe CLP and increase mortality in C57BL/6-KitW-sh/W-sh with severe CLP or infected with Salmonella typhimurium.52
Also, indirect support to our results may come from other mutant mice carrying quantitative or qualitative defects in the MC population. Some of us have recently reported a more severe EAE in histidine decarboxylase (HDC)-deficient mice,28 which are not only genetically unable to produce histamine, but also show paucity in MC number and abnormalities in MC cytoplasmic granules.53 Therefore, it is possible that these mice share with c-Kit mutants the phenotypical defects that depend on MC ablation. It must be noted that some of the results here reported may be associated also to an alteration of histamine signalling in these MC-deficient mice, as histamine has been lately reported to reduce T cell proliferation and IFN-γ production in autoreactive T cells.54
Our data also show some discrepancy with the results obtained by other groups in the KitW-sh/W-sh strain, showing unchanged21 or reduced22 EAE clinical course in these mice in comparison with WT counterpart. The former study highlights that MCs accumulate in the CNS trafficking from the bone marrow during EAE but are dispensable for the development of the disease.21 Interestingly, although in this study no difference in EAE expression between KitW-sh/W-sh and Kit+/+ mice was observed, an increased Ag-specific T-cell proliferation in immunized KitW-sh/W-sh mice was reported, in line with our findings. The latter study shows that milder clinical signs of EAE in KitW-sh/W-sh mice correlate with decreased CD8+ T-cell activation.22 Although the reasons for these discrepancies still need to be determined, we cannot exclude that different mouse housing conditions may affect the outcome of the KitW-sh/W-sh mutation in EAE development, a process dependent on the composition of the gut microflora,55 which in turn may be modulated in a context of MC deficiency.56 Moreover, environmental stress may modulate autoimmunity development in experimental models57 and MCs possess well-known abilities to translate neurological into immunological signals.58
MC-knockin mouse models are useful tools to attribute a specific role to MCs or to a MC-expressed molecule in a particular biological process or pathological condition.29 Indeed, reconstitution of c-Kit mutant mice with in vitro differentiated MCs should recover only MC-dependent defects independently from the kind of c-Kit mutation. Different routes can be used to transfer BMMCs attaining different outcomes. Systemic intravenous injection produced MC accumulation but altered distribution in blood-draining organs with only a modest repopulation at priming sites, ie, the inguinal and axillary lymph node, and in the neighboring skin, according to previous data.29, 39 In particular, the spleen has been recognized to trap most of i.v.-injected MCs, thus inhibiting their spreading to other tissues.39 Thus, intravenous injection of BMMCs may inefficiently reproduce the topography and the quantity of MCs observed in lymphoid organs of Kit+/+ mice in physiological conditions. However, we could detect the rescue of some MC-related biological functions, such as the recovery of normal Treg and granulocyte frequency, in lymph nodes and spleens, respectively, of reconstituted mice. In these conditions, MC transfer failed to recover the normal disease course. Although we cannot exclude that the altered MC distribution at priming sites, especially at draining lymph nodes, might have influenced the clinical outcome of EAE in these reconstituted mice, this result suggests that an immunoregulatory role of MCs in the priming phase of EAE is unlikely. According to previous results in the KitW/W-v strain,18 injected BMMCs failed to repopulate the CNS parenchyma, therefore this experimental setting was not suitable to ascertain whether MCs exerted a regulatory role directly in the inflamed milieu of the CNS. Therefore, tissue-specific MC knockin should be obtained, when possible, by localized MCs injections in order to allow optimal and physiological repopulation. Intravenous or intracranial injection of BMMCs has been recently found to restore the pool of meningeal MCs in KitW/W-v mice, leading to the recovery of a WT-like disease course and to the rescue of neutrophil infiltration in the CNS.20 In our system, although detecting scattered MCs in Kit+/+ mice in the context of the reticulum formed by the arachnoid, the subdural neurothelium, and the dura mater, in line with previous observations,29 we could not find MCs populating the meninges in BMMC-reconstituted KitW-sh/W-sh mice (not shown).
To explore the contribution of immune cells other than MCs in the observed phenotype, we checked the defective frequency of other c-Kit-related cells, such as NK and DCs,45, 46, 47 in KitW-sh/W-sh mice, without finding any difference, according to previous data.29 However, we cannot exclude that DC-expressed c-Kit has some role on immunization, as demonstrated in a model of allergic asthma, in which Ag/adjuvant exposure induced on DCs the expression of both c-Kit and its ligand, promoting Th2 and Th17 responses.45 We then focused our attention on granulocytes because of their reported altered homeostasis in KitW-sh/W-sh mice30 and their involvement in EAE pathogenesis.41 Confirming previous results,30 we found an expanded myeloid compartment in the spleens of KitW-sh/W-sh mice, involving both the Gr1hiCD11bhi and the Gr1int CD11bint subsets. We further characterized these myeloid cells for the Ly6G and Ly6C markers, and found that both subpopulations were expanded in the spleens of KitW-sh/W-sh animals. We looked at the frequency of these cells also in lymph nodes hosting encephalitogenic T-cell priming and found Gr1hiCD11bhi and Ly6G+CD11b+ cells, but not Gr1int CD11bint or Ly6C+CD11b+ cells, selectively expanded by the c-Kit mutation. As the anti-Gr1 mAb used to stain granulocytes reacts with both Ly6G and Ly6C molecules,42 it was not possible to ascertain whether the Ly6C+ entirely corresponded to the Gr1int and the Ly6G+ to the Gr1hi subset. We found that the whole CD11b+ compartment expressed c-Kit, although at a lower level than MCs (not shown), but the c-Kit expression level varied between different subsets and different genotypes, being higher in WT Gr1hiCD11bhi cells compared with mutated Gr1hiCD11bhi, WT Gr1intCD11bint and mutated Gr1intCD11bint cells. These data confirm that, contrary to the KitW/W-v, the KitW-sh/W-sh mutation does not produce an indiscriminate functional knockdown of the c-Kit receptor. Rather, the inversion in a regulatory element upstream the c-Kit gene may finely regulate the effects of c-Kit expression not only in MCs, but also in other cells expressing c-Kit under different transcriptional stimuli. In line with this hypothesis, we found that the Gr1hiCD11bhi compartment seemed more strictly related to the KitW-sh/W-sh mutation than the Gr1intCD11bint counterpart, in terms of peripheral lymphoid accumulation/homing and c-Kit expression level. However, our data also suggest that granulocyte abnormalities detected in KitW-sh/W-sh mice may be ascribed to granulocyte-extrinsic defects, as we could rescue a normal myeloid compartment in the spleens following systemic MC reconstitution. On the contrary, in the KitW/W-v model, the failure to correct peripheral neutropenia with BMMC reconstitution suggests a neutrophil-intrinsic role of the KitW/W-v mutation in shaping the granulocyte compartment.20 It could be proposed that, in KitW-sh/W-sh mice, MCs may actively inhibit granulopoiesis by unknown mechanisms, or passively limit excessive myeloid expansion simply by sequestering the majority of locally available, paracrine or juxtacrine, SCF through the highly expressed c-Kit receptor. This observation suggests to carefully interpret the data obtained in MC-knockin experimental systems, as also non-MC-related defects may be corrected by MC transfer.
We confirmed myeloid cell expansion in the spleen (not shown) and in draining lymph nodes of MOG35–55-immunized mice, involving both Ly6G+ and Ly6C+ compartments. The latter has been recently identified as a monocytic immunosuppressive subset that expanded at EAE onset and inhibited ex vivo T-cell proliferation, thus being attributed to the family of myeloid-derived suppressor cells.59 However, the in vivo suppressive function of Ly6C+ cells in EAE has been questioned by the observation of an attenuated disease development upon Ly6C+ cell pharmacological elimination.60 Conversely, the Ly6G+ fraction was not suppressive ex vivo59 and accordingly Ly6G elimination strongly inhibited the development of both active and passive EAE, suggesting an involvement of granulocytes in the effector phase of EAE.41 Ly6G+ cells, expanded in preimmune KitW-sh/W-sh mice, further increased in lymph nodes on immunization and infiltrated earlier the CNS. We confirmed in vivo the pathogenetic, rather than immunosuppressive, nature of these cells in both c-Kit mutated and WT mice, as both strains showed attenuated and delayed symptoms when treated with anti-Gr1 mAb. However, the alterations carried by KitW-sh/W-sh mice in the Ly6G+ subset were not causally related to the enhanced EAE clinical course, as granulocyte depletion could not abate the difference between KitW-sh/W-sh and Kit+/+ mice in terms of clinical disease expression, despite it dramatically reduced encephalitogenic T-cell responses at the same extent in both groups compared with untreated controls.
Overall our data tend to exclude a role for peripheral MCs in regulating T-cell priming, even though we could detect, in draining lymph nodes of KitW-sh/W-sh mice at d10 on immunization, enhanced T-cell proliferation, increased secretion of proinflammatory cytokines and decreased IL-10 amount, partially in line with previous data.21 The frequency of Th17 and Th1 cells was higher in KitW-sh/W-sh mice at d10 post immunization. Even though the discovery that Th17 cells drove autoimmunity subverted the previous belief that Th1 cells were the most relevant pathogenic cells in EAE, it is now well accepted that both T-helper subsets participates, through distinct mechanisms, to EAE and MS pathogenesis.61 Indeed, the IL-12-Th1 and the IL23-Th17 axes lead to prominent macrophage or granulocyte infiltrate, respectively, in spite of clinically indistinguishable disease courses.40 Th17 encephalitogenic cells displayed the unique feature to enter the not-inflamed CNS62 and to promote the assembly of ectopic lymphoid follicle-like structures in the CNS.63 The observed increase of Th17 and Th1 cells in KitW-sh/W-sh mice may be related to the fact that MCs can exert some influence on T-cell differentiation by releasing a variety of cytokines in response to adjuvants on immunization.64 In the KitW/W-v model, a central role has been attributed to MCs in promoting T-cell priming, but MCs also enhanced inflammation in the effector phase of the disease.18, 38 Indeed, on one side, MC i.v. reconstitution recovered normal EAE, although without repopulating the CNS;18 on the other side, KitW/W-v mice still developed milder symptoms on passive transfer of encephalitogenic T cells.38 Others have found that MCs are attracted from the spleens to the draining lymph nodes upon immunization.18 The cytokine IL-9, produced by Th17 cells in the lymph nodes of MOG35–55-primed mice, has been recently identified to recruit MCs in lymphoid organs and to promote EAE development and progression.65 We have shown in vitro that activated MCs inhibit Treg suppression and promote the differentiation of Th17 effector cells.19 In vivo, MCs colocalized with Th17 cells and Treg in draining lymph nodes of MOG35–55-immunized mice, thus potentially engaging multiple interactions with cells participating to adaptive immune response.19 In this study, we found that immunization in WT mice induced an increase in MC infiltration in draining lymph nodes, which was associated with a preferential distribution of MCs in T-cell perifollicular areas of the lymph node cortex, and with a certain degree of MC clustering. By contrast, no significant change in MC infiltration and distribution was observed in the CNS of WT mice following immunization, supporting that MC functions related to Ag stimulation are mainly played in the DLNs. Analysis of naive and immunized KitW-sh/W-sh mice highlighted the absence of MCs in steady-state and inflamed tissues, respectively. Grippingly, the absence of MCs from DLNs of immunized KitW-sh/W-sh mice was associated with a lower number of IL-10-expressing cells populating DLNs as compared with that observed in WT mice. This was in favor of a role for MCs in regulating IL-10 production during immune response instruction.
A high fraction of degranulated MCs has been detected in the CNS of MBP-immunized mice.66 In a model of allograft tolerance, MC degranulation has been shown to break Treg-mediated immunological tolerance.37 However, by toluidine blue staining, we could not detect significant MC degranulation in draining lymph nodes or in the CNS of MOG35–55-immunized mice. Accordingly, ultrastructural analysis of MCs infiltrating the CNS in a marmoset model of EAE showed selective secretion, rather than degranulation.67
Many data suggest that MCs can engage multiple interactions with Treg.19, 26, 32 In this study, we found a defect in Treg frequency in lymphoid organs of KitW-sh/W-sh mice before and during EAE development, at both acute and chronic phases. The reasons for impaired Treg homeostasis in naive KitW-sh/W-sh mice remain presently obscure. Contrary to another c-Kit mutant, showing severe defects in thymocytes development,68 we could not detect any major alteration in thymocyte subsets in KitW-sh/W-sh mice (data not shown). Notably, BMMC reconstitution restored a physiological Treg frequency in LN of preimmune KitW-sh/W-sh mice. This observation points to a Treg-extrinsic role of this c-Kit mutation in shaping Treg compartment and suggests that MCs may not only mold Treg plasticity but also support Treg homeostasis. The OX40–OX40L axis may be involved in a defective Treg expansion in MC-deficient mice. Indeed, MCs constitutively express OX40L26 and OX40 is a key signal in supporting Treg fitness especially under inflammatory conditions.69 Treg are important suppressor cells in both the initiation and the recovery phase of EAE,70 therefore the enhanced EAE disease course in KitW-sh/W-sh mice could be associated to the decreased Treg accumulation in LN and spleen. However, Kit+/+ and KitW-sh/W-sh mice showed the same proportion of CNS-infiltrating Treg and BMMCs-reconstituted mice still developed exacerbated symptoms despite the recovery of a physiological Treg frequency.
MCs residing and/or recruited to the CNS has several roles in non-immunological conditions. KitW-sh/W-sh mice show a greater anxiety-like phenotype than WT controls.71 Located in close proximity with the blood–brain barrier, MCs represent early responders to cerebral ischemia and potent inflammatory effector cells.72 MCs may even modulate tumor metastasization into the brain.73 In KitW/W-v, meningeal MCs promote a breach in the blood–brain barrier to allow the entry of inflammatory cells.20 Overall these functions imply a role for MCs in increasing, rather than regulating, the permeability of blood-brain barrier. On the contrary, we found here an enhanced immune infiltrate in MC-deficient KitW-sh/W-sh mice, especially granulocytes. Of note, we found more granulocytes infiltrating also the CNS of HDC−/− mice, carrying histamine deficiency and MC paucity.28 Future experiments will unravel the true role of MCs in promoting or regulating the access of immune cells to inflamed CNS.
References
Galli SJ, Nakae S, Tsai M . Mast cells in the development of adaptive immune responses. Nat Immunol 2005;6:135–142.
Kalesnikoff J, Galli SJ . New developments in mast cell biology. Nat Immunol 2008;9:1215–1223.
Galli SJ, Grimbaldeston M, Tsai M . Immunomodulatory mast cells: negative, as well as positive, regulators of immunity. Nat Rev 2008;8:478–486.
Kawakami T . A crucial door to the mast cell mystery knocked in. J Immunol 2009;183:6861–6862.
Chen R, Ning G, Zhao ML, et al. Mast cells play a key role in neutrophil recruitment in experimental bullous pemphigoid. J Clin Invest 2001;108:1151–1158.
Lee DM, Friend DS, Gurish MF, et al. Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science 2002;297:1689–1692.
Hochegger K, Siebenhaar F, Vielhauer V, et al. Role of mast cells in experimental anti-glomerular basement membrane glomerulonephritis. Eur J Immunol 2005;35:3074–3082.
Kanamaru Y, Scandiuzzi L, Essig M, et al. Mast cell-mediated remodeling and fibrinolytic activity protect against fatal glomerulonephritis. J Immunol 2006;176:5607–5615.
Zhou JS, Xing W, Friend DS, et al. Mast cell deficiency in Kit(W-sh) mice does not impair antibody-mediated arthritis. J Exp Med 2007;204:2797–2802.
Pedotti R, De Voss JJ, Steinman L, et al. Involvement of both ‘allergic’ and ‘autoimmune’ mechanisms in EAE, MS and other autoimmune diseases. Trends Immunol 2003;24:479–484.
Ibrahim MZ, Reder AT, Lawand R, et al. The mast cells of the multiple sclerosis brain. J Neuroimmunol 1996;70:131–138.
Olsson Y . Mast cells in plaques of multiple sclerosis. Acta Neurol Scand 1974;50:611–618.
Lock C, Hermans G, Pedotti R, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med 2002;8:500–508.
Couturier N, Zappulla JP, Lauwers-Cances V, et al. Mast cell transcripts are increased within and outside multiple sclerosis lesions. J Neuroimmunol 2008;195:176–185.
Rozniecki JJ, Hauser SL, Stein M, et al. Elevated mast cell tryptase in cerebrospinal fluid of multiple sclerosis patients. Ann Neurol 1995;37:63–66.
Secor VH, Secor WE, Gutekunst CA, et al. Mast cells are essential for early onset and severe disease in a murine model of multiple sclerosis. J Exp Med 2000;191:813–822.
Robbie-Ryan M, Tanzola MB, Secor VH, et al. Cutting edge: both activating and inhibitory Fc receptors expressed on mast cells regulate experimental allergic encephalomyelitis disease severity. J Immunol 2003;170:1630–1634.
Tanzola MB, Robbie-Ryan M, Gutekunst CA, et al. Mast cells exert effects outside the central nervous system to influence experimental allergic encephalomyelitis disease course. J Immunol 2003;171:4385–4391.
Piconese S, Gri G, Tripodo C, et al. Mast cells counteract regulatory T-cell suppression through interleukin-6 and OX40/OX40L axis toward Th17-cell differentiation. Blood 2009;114:2639–2648.
Sayed BA, Christy AL, Walker ME, et al. Meningeal mast cells affect early T cell central nervous system infiltration and blood-brain barrier integrity through TNF: a role for neutrophil recruitment? J Immunol 2010;184:6891–6900.
Bennett JL, Blanchet MR, Zhao L, et al. Bone marrow-derived mast cells accumulate in the central nervous system during inflammation but are dispensable for experimental autoimmune encephalomyelitis pathogenesis. J Immunol 2009;182:5507–5514.
Stelekati E, Bahri R, D’Orlando O, et al. Mast cell-mediated antigen presentation regulates CD8+ T cell effector functions. Immunity 2009;31:665–676.
Williams CM, Galli SJ . Mast cells can amplify airway reactivity and features of chronic inflammation in an asthma model in mice. J Exp Med 2000;192:455–462.
Norman MU, Hwang J, Hulliger S, et al. Mast cells regulate the magnitude and the cytokine microenvironment of the contact hypersensitivity response. Am J Pathol 2008;172:1638–1649.
Yang K, Vega JL, Hadzipasic M, et al. Deficiency of thrombospondin-1 reduces Th17 differentiation and attenuates experimental autoimmune encephalomyelitis. J Autoimmun 2009;32:94–103.
Gri G, Piconese S, Frossi B, et al. CD4+CD25+ regulatory T cells suppress mast cell degranulation and allergic responses through OX40-OX40L interaction. Immunity 2008;29:771–781.
Piddlesden SJ, Storch MK, Hibbs M, et al. Soluble recombinant complement receptor 1 inhibits inflammation and demyelination in antibody-mediated demyelinating experimental allergic encephalomyelitis. J Immunol 1994;152:5477–5484.
Musio S, Gallo B, Scabeni S, et al. A key regulatory role for histamine in experimental autoimmune encephalomyelitis: disease exacerbation in histidine decarboxylase-deficient mice. J Immunol 2006;176:17–26.
Grimbaldeston MA, Chen CC, Piliponsky AM, et al. Mast cell-deficient W-sash c-kit mutant Kit W-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am J Pathol 2005;167:835–848.
Nigrovic PA, Gray DH, Jones T, et al. Genetic inversion in mast cell-deficient (W(sh)) mice interrupts corin and manifests as hematopoietic and cardiac aberrancy. Am J Pathol 2008;173:1693–1701.
Mendel I, Kerlero de Rosbo N, Ben-Nun A . A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: fine specificity and T cell receptor V beta expression of encephalitogenic T cells. Eur J Immunol 1995;25:1951–1959.
Lu LF, Lind EF, Gondek DC, et al. Mast cells are essential intermediaries in regulatory T-cell tolerance. Nature 2006;442:997–1002.
Grimbaldeston MA, Nakae S, Kalesnikoff J, et al. Mast cell-derived interleukin 10 limits skin pathology in contact dermatitis and chronic irradiation with ultraviolet B. Nat Immunol 2007;8:1095–1104.
Villa I, Skokos D, Tkaczyk C, et al. Capacity of mouse mast cells to prime T cells and to induce specific antibody responses in vivo. Immunology 2001;102:165–172.
Yoshikawa T, Imada T, Nakakubo H, et al. Rat mast cell protease-I enhances immunoglobulin E production by mouse B cells stimulated with interleukin-4. Immunology 2001;104:333–340.
Gounaris E, Blatner NR, Dennis K, et al. T-regulatory cells shift from a protective anti-inflammatory to a cancer-promoting proinflammatory phenotype in polyposis. Cancer Res 2009;69:5490–5497.
de Vries VC, Wasiuk A, Bennett KA, et al. Mast cell degranulation breaks peripheral tolerance. Am J Transplant 2009;9:2270–2280.
Gregory GD, Robbie-Ryan M, Secor VH, et al. Mast cells are required for optimal autoreactive T cell responses in a murine model of multiple sclerosis. Eur J Immunol 2005;35:3478–3486.
Wolters PJ, Mallen-St Clair J, Lewis CC, et al. Tissue-selective mast cell reconstitution and differential lung gene expression in mast cell-deficient Kit(W-sh)/Kit(W-sh) sash mice. Clin Exp Allergy 2005;35:82–88.
Kroenke MA, Carlson TJ, Andjelkovic AV, et al. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med 2008;205:1535–1541.
McColl SR, Staykova MA, Wozniak A, et al. Treatment with anti-granulocyte antibodies inhibits the effector phase of experimental autoimmune encephalomyelitis. J Immunol 1998;161:6421–6426.
Fleming TJ, Fleming ML, Malek TR . Selective expression of Ly-6G on myeloid lineage in mouse bone marrow: RB6-8C5 mAb to granulocyte-differentiation antigen (Gr-1) detects members of the Ly-6 family. J Immunol 1993;151:2399–2404.
Mildner A, Djukic M, Garbe D, et al. Ly-6G+CCR2− myeloid cells rather than Ly-6ChighCCR2+ monocytes are required for the control of bacterial infection in the central nervous system. J Immunol 2008;181:2713–2722.
Pittoni P, Piconese S, Tripodo C, et al. Tumor-intrinsic and extrinsic roles of c-Kit in cancer: mast cells as the primary off-target of tyrosine kinase inhibitors. Oncogene; advance online publication, 8 November 2010; doi:10.1038/onc.2010.494.
Krishnamoorthy N, Oriss TB, Paglia M, et al. Activation of c-Kit in dendritic cells regulates T helper cell differentiation and allergic asthma. Nat Med 2008;14:565–573.
Borg C, Terme M, Taieb J, et al. Novel mode of action of c-kit tyrosine kinase inhibitors leading to NK cell-dependent antitumor effects. J Clin Invest 2004;114:379–388.
Taieb J, Chaput N, Menard C, et al. A novel dendritic cell subset involved in tumor immunosurveillance. Nat Med 2006;12:214–219.
Stromnes IM, Goverman JM . Active induction of experimental allergic encephalomyelitis. Nat Protoc 2006;1:1810–1819.
Chan JC, Knudson O, Wu F, et al. Hypertension in mice lacking the proatrial natriuretic peptide convertase corin. Proc Natl Acad Sci USA 2005;102:785–790.
Platten M, Youssef S, Hur EM, et al. Blocking angiotensin-converting enzyme induces potent regulatory T cells and modulates TH1- and TH17-mediated autoimmunity. Proc Natl Acad Sci USA 2009;106:14948–14953.
Shelley O, Murphy T, Lederer JA, et al. Mast cells and resistance to peritoneal sepsis after burn injury. Shock 2003;19:513–518.
Piliponsky AM, Chen CC, Grimbaldeston MA, et al. Mast cell-derived TNF can exacerbate mortality during severe bacterial infections in C57BL/6-KitW-sh/W-sh mice. Am J Pathol 2010;176:926–938.
Ohtsu H, Tanaka S, Terui T, et al. Mice lacking histidine decarboxylase exhibit abnormal mast cells. FEBS Lett 2001;502:53–56.
Lapilla M, Gallo B, Martinello M, et al. Histamine regulates autoreactive T cell activation and adhesiveness in inflamed brain microcirculation. J Leukoc Biol 2011;89:259–267.
Yokote H, Miyake S, Croxford JL, et al. NKT cell-dependent amelioration of a mouse model of multiple sclerosis by altering gut flora. Am J Pathol 2008;173:1714–1723.
Bischoff SC . Physiological and pathophysiological functions of intestinal mast cells. Semin Immunopathol 2009;31:185–205.
Poliak S, Mor F, Conlon P, et al. Stress and autoimmunity: the neuropeptides corticotropin-releasing factor and urocortin suppress encephalomyelitis via effects on both the hypothalamic-pituitary-adrenal axis and the immune system. J Immunol 1997;158:5751–5756.
Theoharides TC, Konstantinidou AD . Corticotropin-releasing hormone and the blood-brain-barrier. Front Biosci 2007;12:1615–1628.
Zhu B, Bando Y, Xiao S, et al. CD11b+Ly-6C(hi) suppressive monocytes in experimental autoimmune encephalomyelitis. J Immunol 2007;179:5228–5237.
King IL, Dickendesher TL, Segal BM . Circulating Ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood 2009;113:3190–3197.
Steinman L . A rush to judgment on Th17. J Exp Med 2008;205:1517–1522.
Reboldi A, Coisne C, Baumjohann D, et al. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol 2009;10:514–523.
Jager A, Dardalhon V, Sobel RA, et al. Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J Immunol 2009;183:7169–7177.
Sayed BA, Christy A, Quirion MR, et al. The master switch: the role of mast cells in autoimmunity and tolerance. Ann Rev Immunol 2008;26:705–739.
Nowak EC, Weaver CT, Turner H, et al. IL-9 as a mediator of Th17-driven inflammatory disease. J Exp Med 2009;206:1653–1660.
Brenner T, Soffer D, Shalit M, et al. Mast cells in experimental allergic encephalomyelitis: characterization, distribution in the CNS and in vitro activation by myelin basic protein and neuropeptides. J Neurol Sci 1994;122:210–213.
Letourneau R, Rozniecki JJ, Dimitriadou V, et al. Ultrastructural evidence of brain mast cell activation without degranulation in monkey experimental allergic encephalomyelitis. J Neuroimmunol 2003;145:18–26.
Waskow C, Rodewald HR . Lymphocyte development in neonatal and adult c-Kit-deficient (c-KitW/W) mice. Adv Exp Med Biol 2002;512:1–10.
Piconese S, Pittoni P, Burocchi A, et al. A non-redundant role for OX40 in the competitive fitness of Treg in response to IL-2. Eur J Immunol 2010;40:2902–2913.
O’Connor RA, Anderton SM . Foxp3+ regulatory T cells in the control of experimental CNS autoimmune disease. J Neuroimmunol 2008;193:1–11.
Nautiyal KM, Ribeiro AC, Pfaff DW, et al. Brain mast cells link the immune system to anxiety-like behavior. Proc Natl Acad Sci USA 2008;105:18053–18057.
Lindsberg PJ, Strbian D, Karjalainen-Lindsberg ML . Mast cells as early responders in the regulation of acute blood-brain barrier changes after cerebral ischemia and hemorrhage. J Cereb Blood Flow Metab 2010;30:689–702.
Theoharides TC, Rozniecki JJ, Sahagian G, et al. Impact of stress and mast cells on brain metastases. J Neuroimmunol 2008;205:1–7.
Acknowledgements
We thank Stephen J Galli, Carlo E Pucillo and Barbara Frossi for their helpful discussion, and Lawrence Steinman for conducting some of these experiments in his laboratory. We acknowledge Ivano Arioli for technical assistance. This work was supported by grants from the Italian Ministry of Health (to MPC and Ricerca Finalizzata 2007 to RP), The National Multiple Sclerosis Society New York (to RP), Fondazione Italiana Sclerosi Multipla-FISM 2009/R/20 (to RP) and Associazione Italiana Ricerca sul Cancro (to MPC). SP is supported by My First AIRC grant (8726). MC is supported by a training (research) fellowship (2008/B/2) from the Fondazione Italiana Sclerosi Multipla. PP is supported by a fellowship from Fondazione Italiana Ricerca sul Cancro.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
Supplementary information
Rights and permissions
About this article
Cite this article
Piconese, S., Costanza, M., Musio, S. et al. Exacerbated experimental autoimmune encephalomyelitis in mast-cell-deficient KitW-sh/W-sh mice. Lab Invest 91, 627–641 (2011). https://doi.org/10.1038/labinvest.2011.3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2011.3
Keywords
This article is cited by
-
Mast cells and angiogenesis in multiple sclerosis
Inflammation Research (2020)
-
Role of Mast Cells in the Pathogenesis of Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis
Clinical Reviews in Allergy & Immunology (2017)
-
Immune mediators of chronic pelvic pain syndrome
Nature Reviews Urology (2014)
-
Focal brain inflammation and autism
Journal of Neuroinflammation (2013)
-
The role of mast cells in neuroinflammation
Acta Neuropathologica (2013)
