
Abstract
Glomerular parietal epithelial cells (PECs) are precursors to podocytes in mature glomeruli; however, as progenitors, the distinct intrinsic mechanisms that allow for repeated periods of cell-cycle arrest and re-entry of PECs after glomerulogenesis are unknown. Here, we show that the Src-suppressed protein kinase C substrate (SSeCKS), a multivalent scaffolding A kinase anchoring protein, sequesters cyclin D1 in the cytoplasm of quiescent PECs. SSeCKS expression is induced in embryonic PECs, but not in embryonic podocytes, starting at the S phase of glomerulogenesis, and is constitutively expressed postnatally by PECs, but not podocytes, in normal glomeruli. Cyclin D1 was immunoprecipitated with SSeCKS from capsulated glomeruli containing PECs, whereas decapsulated glomeruli without PECs lacked SSeCKS and cyclin D1. Cell–cell contact inhibition of proliferation in cultured PECs induced SSeCKS expression and binding of cyclin D1 by SSeCKS in the cytoplasm, whereas phosphorylation of SSeCKS by activated protein kinase C disrupted binding, resulting in nuclear translocation of cyclin D1. SSeCKS−/− mice showed hyperplasia of PECs in otherwise normal glomeruli and developed significantly worse proteinuric glomerular disease, marked by increased PEC proliferation and expression of nuclear cyclin D1, from nephrotoxic nephritis. These results suggest that SSeCKS controls the localization and activity of cyclin D1 in PECs and influences proliferative injury in the glomerulus.
Similar content being viewed by others


Main
Glomerular visceral epithelial cells (podocytes) were considered irreplaceble until the discovery that contiguous glomerular parietal epithelial cells (PECs) are progenitors for nascent podocytes during postnatal life.1, 2 Coined a ‘renopoietic system’ for podocytes, PECs in normal glomeruli comprise a population of renal tissue stem cells along the urinary pole of Bowman's basement membrane (BBM) and adjacent daughter cells showing increased speciation directed towards the vascular pole of BBM for the podocyte lineage.1, 2 At steady state, the proliferation and migration of PECs to replace senesced podocytes likely occurs without appreciable loss of glomerular function, whereas abberant hyperplastic or hypoplastic repair by PECs appears to contribute to the formation of proliferative and cytopenic podocytopathies, respectively.3, 4, 5 This emerging role for PECs in glomerular health and disease raises the possibility for manipulating the proliferative response of PECs for therapeutic benefit.
As precursors for podocytes in postnatal glomeruli, the distinct intrinsic mechanisms that allow for repeated periods of cell-cycle arrest and re-entry of PECs apart from podocytes are unknown.6 Unlike the proliferative independence of embryonic stem cells from G1-to-S checkpoint control by retinoblastoma protein (Rb), research on resting progenitors in other postnatal tissues suggests that mitogen-induced cyclin D1-cyclin-dependent kinase 4/6 phosphorylation of Rb drives their cell-cycle progression.6 Interestingly, during the late stages of glomerulogenesis, proliferating embryonic podocytes downregulate cyclin D1 and upregulate cyclin D3 and the G1-to-S cell-cycle inhibitors, p27 and p57, expression that remains constitutive in mature podocytes in postnatal glomeruli.7, 8 This terminal cell-cycle state for differentiated podocytes is not evident for PECs, consistent with the latter's function as contiguous progenitors.1, 2 However, the cell-cycle mechanisms that distinctly maintain the resting state and proliferative potential of PECs are unknown.
Here, we hypothesize that Src-suppressed protein kinase C substrate (SSeCKS),9 encoded by the rodent orthologue of human gravin/akap12, sequesters cyclin D1 in mouse PECs, serving as a gating checkpoint for cell-cycle arrest. SSeCKS is a cytoplasmic, multivalent scaffolding protein in the family of A kinase anchoring proteins (AKAPs)10 that was originally cloned as a tumor suppressor.11, 12 SSeCKS is unique among AKAPs by containing protein kinase C (PKC) phosphorylation sites that flank cyclin-binding motifs, which bind cyclin D1.13, 14 In response to cell–cell contact inhibition of proliferation,15 SSeCKS expression is induced, resulting in the sequestration of existing cyclin D1 protein in the cytoplasm and the downregulation of further cyclin D1 expression through extracellular signal-regulated kinase-dependent pathways. This leads to growth arrest by inhibiting G1-to-S cell-cycle progression.13, 16 Phosphorylation of SSeCKS by activated PKC (aPKC), signaling induced by many mitogens,17 releases this binding of cyclin D1, allowing its nuclear translocation and G1-to-S cell-cycle progression.13, 14 In previous studies, we discovered that SSeCKS is highly expressed by mature PECs, but is not detected in mature podocytes, in postnatal glomeruli.18 This differential expression raises the possibility that SSeCKS is distinctly involved in maintaining the resting state and proliferative potential of PECs apart from podocytes by regulating cyclin D1.
MATERIALS AND METHODS
Mice
All experiments in mice were approved by Institutional Animal Care and Use Committee. Archival formalin-fixed, paraffin-embedded kidneys from pre-natal mice at embryonic day 18 (E18), cyclin D1−/− mice, diseased Tg26 mice (Tg26), diseased mice with nephrotoxic nephritis (NTN), diseased rats with passive Heymann nephritis (PHN), diseased rats with puromycin aminonucleoside nephrosis (PAN), and diseased mice with adriamycin nephrosis (AN), and the development of C57Bl/6 SSeCKS−/− and SSeCKS+/+ (wild-type) mice, are described previously.7, 19, 20, 21, 22 Kidneys from male SSeCKS+/+ and male SSeCKS−/− mice at 15 weeks of age were fixed in Karnovsky's solution for imaging glomerular ultrastructure by the University of Washington Pathology Research Service Laboratory on a Tecnai G2 Spirit Bio Twin Electron Microscope. Magnetic-bead isolation of capsulated and decapsulated glomeruli from wild-type mice following differential size sieving of digested kidneys was performed as described previously,23 with isolated glomeruli immediately lysed in RIPA buffer (Teknova, Hollister, CA, USA) containing Complete Protease Inhibitor (Roche, Indianapolis, IN, USA), 50 mM sodium fluoride and 0.1 mM sodium orthovanadate at 4°C to collect total protein. Passive NTN24 was induced in cohorts of male SSeCKS+/+ mice (n=5) and male SSeCKS−/− mice (n=5) at 15 weeks of age by intraperitoneal injection of nephrotoxic immunoglobulin (12.5 mg/20 g body weight), or an equivalent volume of vehicle (1 × PBS, pH 7.4), on 2 consecutive days. Urine was collected from each mouse on day 14 after the second intraperitoneal injection immediately before collection, formalin fixation and paraffin embedding of kidneys. Urine was assayed for the concentration of albumin and creatinine using the Albuwell Murine Microalbumin ELISA (Exocell, Philadelphia, PA, USA) and Creatinine Colorimetric Microplate Assay (Cayman Chemical, Ann Arbor, MI, USA), respectively, as per the manufacturer's protocols.
Tissue Culture
Mouse PECs were cultured as described previously.23, 25 To study cell–cell contact inhibition in PECs, cells growing under conditions permissive for proliferation (33° with IFN-γ) were analyzed at 50, 80, and 100% confluency, or following differentiation at 100% confluency for 14 days under non-permissive conditions (37°C without IFN-γ). Total protein from cells was prepared by lysing cells in RIPA buffer containing Complete Protease inhibitor, 50 mM sodium fluoride, and 0.1 mM sodium orthovanadate at 4°C. Cytoplasmic protein from cells was prepared by lysing cells in fractionation buffer (10 mM HEPES, 50 mM NaCl, 0.5 M sucrose, 0.1 mM EDTA, 0.5% Triton X-100, 1 mM DTT, Complete Protease Inhibitor, 50 mM sodium fluoride, 0.1 mM sodium orthovanadate, pH 7.9) and collecting supernatant following the pelleting of nuclei at 3000 r.p.m. for 10 min at 4°C. PKC was activated in differentiated PECs using 100 mM phorbol 12,13-diacetate (Sigma Aldrich, St Louis, MO, USA) or 10 μg/ml soluble TNF-α (R&D Systems, Minneapolis, MN, USA), or inhibited by pre-treating cells for 2 h with staurosporine aglycone (Sigma Aldrich) before the addition of activators.
Immunofluorescence
Immunofluorescence detection of SSeCKS, phospho-SSeCKS ser283, and cyclin D1 in cultured PECs was performed as described previously.18 Immunofluorescence detection of SSeCKS and cyclin D1 in 4 μm kidneys sections was performed after sections were cleared of paraffin (Histoclear; National Diagnostics, Atlanta, GA, USA), rehydrated, retrieved for antigen by microwave boiling in 10 mM citrate (pH 6.0), and blocked with 5% bovine serum albumin. Primary antibodies were the following: rabbit polyclonal anti-SSeCKS (Irwin Gelman Laboratory); rabbit polyclonal anti-phospho-SSeCKS ser283 (Irwin Gelman Laboratory); and mouse monoclonal anti-cyclin D1 (Millipore, Temecula, CA, USA). Secondary antibodies were the following: Alexa 488-conjugated anti-rabbit IgG (Invitrogen, San Diego, CA, USA) and TXR-conjugated anti-mouse IgG (Invitrogen). Nuclei were stained with Hoechst 33342 (Thermo Scientific, Natick, MA, USA). Warp Red Chromogen Kit (BioCare Medical, Concord, CA, USA) was also used for secondary detection of cyclin D1 by white light and fluorescence in kidney sections without additional stains. Imaging was performed on a DMRB Fluorescence Microscope (Leica, Richmond, IL, USA).
Western Blotting
Protein lysates where separated by SDS-PAGE and transferred to PDVF membranes (Millipore) for immunoblotting. Total protein from isolated glomeruli was immunoblotted for SSeCKS (1:200), cyclin D1 (1:500), cyclin D3 (1:500; mouse monoclonal; Lab Vision, Fremont, CA, USA), and PAX8 (1:500; rabbit polyclonal; ProteinTech Group, Chicago, IL, USA), using tubulin-α (1:500; mouse monoclonal mouse; Lab Vision) as a loading control. Total protein from differentiated PECs with activated or inhibited PKC was immunoblotted for phospho-SSeCKS ser283 (1:200), using SSeCKS as its own loading reference. For immunoprecipitation of cyclin D1 with SSeCKS, 100 μg of protein from capsulated glomeruli or from cultured, differentiated PECs was incubated with rabbit polyclonal anti-SSeCKS antibody overnight at 4°C, followed by binding to protein A/G PLUS-Agarose (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for SDS-PAGE separation of cyclin D1 and SSeCKS. The reverse immunoprecipitation of SSeCKS using cyclin D1 as bait was also performed on cultured PECS. Primary antibodies were detected using HRP-conjugated antibody-activated chemiluminescence (Amersham, Piscataway, NJ, USA) imaged on a ChemiDoc EQ system (BioRad Laboratories, Berkeley, CA, USA). Western blots shown are representative of three separate experiments.
Quantitative Histopathology
The extent of cellular lesions in the glomeruli of each mouse with NTN was quantified by renal pathologists of the University of Washington Pathology Research Service Laboratory naive to the experimental protocol. Periodic acid Schiff-stained coronal kidney sections were used to calculate the percent of total glomeruli with any cellular lesion. Following the preparation of 4 μm kidney sections as described above, immunohistochemical staining of PAX8, cyclin D1, or Wilms' tumor antigen (WT1; Santa Cruz Biotechnology) in kidneys was by secondary detection using biotinylated antibodies and ABC-Elite (Vector Laboratories, Burlingame, CA, USA) system, as per the manufacturer's protocol. The average number of PECs per glomerulus in normal SSeCKS+/+ mice (n=5) and normal SSeCKS−/− mice (n=5) was calculated by counting PAX8-positive nuclei around the circumference of 20 equivalent glomeruli per mouse to determine the average PEC diameter (diameter=2r), then dividing the glomerular sphere surface area (area=4πr2) by the circular PEC surface area (area=πr2). The average number of podocytes per glomerulus in normal SSeCKS+/+ mice and normal SSeCKS−/− mice was calculated by counting the number of WT1-positive nuclei in 10 glomeruli per mouse. The number of PAX8-positive nuclei or cyclin D1-positive nuclei per diseased glomerulus in each mouse with NTN was calculated by dividing the number of positive-stained nuclei in all glomeruli with cellular lesions by the number of glomeruli with cellular lesions in coronal kidney section.
Statistics
All values are expressed as mean±s.d. Statistical significance was tested by the two-tailed t-test. Significance was tested at the 0.05 level of probability.
RESULTS
SSeCKS and Cyclin D1 in PECs during and after Glomerulogenesis
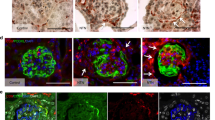
During glomerulogenesis, embryonic podocytes but not embryonic PECs constitutively express p27 and p57 starting at the capillary-loop stage and continuing on into postnatal life, marking their cell-cycle exit as terminally differentiated constituents of the glomerular filtration barrier (GFB).8 However, the developmental program that separately establishes the resting proliferative potential of PECs as progenitors in postnatal kidneys is unknown. Thus, we studied the expression of SSeCKS by immunofluorescence (IF) in pre-natal mouse kidneys at E18, which exhibits all stages of glomerulogenesis. SSeCKS was not detected in embryonic cells forming renal vesicles, ureteric buds, or comma-shaped bodies of primordial glomeruli (Figure 1a and b). By the S-stage, however, SSeCKS was detected in the cytoplasm of filamentous embryonic cells forming the parietal surface of S-shaped bodies (Figure 1c), cells destined to become PECs.1

Expression and interaction of Src-suppressed protein kinase C substrate (SSeCKS) and cyclin D1 during and after glomerulogenesis. Insets in the lower right of panels outline the anatomy shown by fluorescence microscopy, in which nuclei stain blue, SSeCKS stains green, and cyclin D1 stains red. (a) SSeCKS is not detected in embryonic cells forming renal vesicles or ureteric buds. (b) SSeCKS is not detected in embryonic cells forming comma-shaped bodies. (c) SSeCKS is expressed by embryonic parietal epithelial cells (PECs) forming the parietal surface of S-shaped bodies. (d) Cyclin D1 is expressed in the nuclei of SSeCKS-negative embryonic podocytes forming the visceral surface of S-shaped bodies (arrows), adjacent to SSeCKS-positive embryonic PECs. (e) SSeCKS is expressed by embryonic PECs at the capillary-loop stage. (f) Cyclin D1 is not detected in SSeCKS-negative embryonic podocytes or in SSeCKS-positive embryonic PECs at the capillary-loop stage. (g) SSeCKS is expressed by embryonic PECs in fully-formed glomeruli. (h) The expression of SSeCKS is limited to PECs demarcated by the transition zones with contiguous epithelial cells at the vascular (arrows) and urinary (arrowheads) poles of Bowman's basement membrane (BBM). (i) Western immunoblots of protein lysates from capsulated and decapsulated glomeruli isolated from separate mice showing that SSeCKS and cyclin D1 are not abundant in decapsulated glomeruli lacking paired box gene 8 (PAX8)-positive PECs, but retaining cyclin D3-positive podocytes. (j) Western immunoblots (IB) showing binding of cyclin D1 to SSeCKS immunoprecipitated (IP) from protein lysates of capsulated glomeruli isolated from separate mice. Magnifications: × 400 in panels a, b, c, e, f, g, and h; × 2000 in panel d.
This distinct expression of SSeCKS at the S-stage in embryonic PECs, but not in embryonic podocytes, was also present at the capillary-loop stage (Figure 1e) and in PECs in fully formed glomeruli (Figure 1g), the latter shown previously in postnatal kidneys.18 The delimited expression of SSeCKS by mature PECs was evident at the transition zones between PECs and other contiguous epithelial cells at the urinary and vascular poles of BBM (Figure 1h). Co-staining for cyclin D1 by IF showed that nuclear cyclin D1 was present in SSeCKS-negative embryonic podocytes forming the visceral surface of S-shaped bodies (Figure 1d), as shown previously.7 However, cyclin D1 was not readily detected in the nucleus or cytoplasm of filamentous, SSeCKS-positive embryonic PECs at the S-stage, nor during the capillary-loop stage when embryonic podocytes no longer express cyclin D1 (Figure 1f) and begin to constitutively express cyclin D3.7 To address whether this lack of co-staining within PECs reflects the limits of sensitivity or masking of our IF to detect cytoplasmic cyclin D1 in low abundance in thin tissue sections, we single stained with a signal amplifying red chromogen detectable by white light and fluorescence, using kidneys from cyclin D1−/− mice as a negative control.7 Cyclin D1 was detected in the cytoplasm of normal PECs, suggesting its sequestration by SSeCKS, whereas PECs in cyclin D1−/− mice showed no staining for cyclin D1 (Supplementary Figure 1).
We confirmed these suggestive findings from IF by immunoprecipitating cyclin D1 with SSeCKS from isolated glomeruli. Only capsulated glomeruli containing PECs, marked by positivity for paired box gene 8 (PAX8),25 expressed abundant SSeCKS and cyclin D1 (Figure 1i). In comparison, decapsulated glomeruli lacking PECs, but retaining cyclin D3-positive podocytes, did not express significant cyclin D1 or SSeCKS. This differentially expressed cyclin D1 present in capsulated glomeruli immunoprecipitated with SSeCKS (Figure 1j). Taken together, these results suggest that SSeCKS may be distinctly involved in maintaining the resting state of PECs apart from podocytes by binding and sequestering a stable pool of cyclin D1, mechanisms that initiate during glomerulogenesis.
SSeCKS and Cyclin D1 in Cultured PECs
To further examine the control of cyclin D1 by SSeCKS in PECs, we studied the expression and binding interaction of cyclin D1 and SSeCKS in cultured PECs.23 Increasing cell–cell contact between PECs from 50 to 100% cell confluency induced SSeCKS expression in the cytoplasm, concomitant with the redistribution of cyclin D1 from nuclear to cytoplasmic compartments and marked downregulation of total cyclin D1 expression (Figure 2a and b), consistent with previous data.13, 14, 15, 16 This cytoplasmic cyclin D1 immunoprecipitated with SSeCKS (Figure 2b and Supplementary Figure 2), including after differentiation of fully confluent PECs over 14 days to mimic the state of mature PECs in normal glomeruli,23 corroborating our findings of this binding interaction between SSeCKS and cyclin D1 at steady-state in vivo.

Expression and interaction of Src-suppressed protein kinase C substrate (SSeCKS) and cyclin D1 in cultured parietal epithelial cells (PECs). (a) Fluorescence detection of SSeCKS (green), cyclin D1 (red), and nuclei (blue) during increasing cell–cell contact between PECs from 50 to 100% cell confluency showing induction of SSeCKS in the cytoplasm with concomitant nuclear to cytoplasmic redistribution of cyclin D1, compartmentalization that remains in fully differentiated (FD) PECs. (b) Western immunoblots (IB) showing downregulation of cyclin D1 and its immunoprecipitation (IP) with cytoplasmic SSeCKS during increasing cell–cell contact between PECs from 50 to 100% cell confluency, binding that remains in fully differentiated PECs.
SSeCKS is a substrate for aPKC,12 and phosphorylation of SSeCKS by aPKC releases its scaffolding function, thereby allowing cytoplasmic-to-nuclear translocation of cyclin D1.13, 14 Using a monoclonal antibody that specifically detects phosphorylation of serine 283 in SSeCKS, a target residue for aPKC,12 we detected rapid phosphorylation of SSeCKS in PECs following activation of PKC by the phorbol ester, phorbol 12,13-diacetate, or by soluble TNF-α, the latter a known PKC activator26 and inflammatory mitogen in the proliferative podocytopathies (Figure 3a).27, 28 This phosphorylation was abrogated by the pan-PKC inhibitor, staurosporine aglycone. The immediate-early upregulation of SSeCKS expression that occurs with mitogenic signaling is also evident in PECs.11, 16 As a separate control, mouse fibroblasts expressing SSeCKS also showed phosphorylation at serine 283 following activation of PKC (Figure 3b). Cyclin D1 redistributed from the cytoplasm to the nucleus in PECs with phosphorylation of SSeCKS by aPKC (Figure 3c). Taken together, these results suggest that SSeCKS stably sequesters cyclin D1 in the cytoplasm of PECs during growth arrest in cell–cell contact, with loss of this compartmentalization of cyclin D1 following phosphorylation of SSeCKS by aPKC.

Phosphorylation of Src-suppressed protein kinase C substrate (SSeCKS) by activated protein kinase C (aPKC) in cultured parietal epithelial cells (PECs). (a) Western immunoblots showing rapid phosphorylation of SSeCKS at serine 283 (phospho-SSeCKS ser283) within 5 min of activation of PKC in PECs by phorbol 12,13-diacetate (PDA) or tumor necrosis factor-α (TNF-α), signaling abrogated by staurosporine aglycone (inhibitor). (b) Control western immunoblots showing the same signaling as in panel a in mouse fibroblasts expressing SSeCKS. (c) Fluorescence detection of phospho-SSeCKS ser283 (green), cyclin D1 (red), and nuclei (blue) in fully differentiated PECs showing phosphorylation of SSeCKs and cytoplasmic-to-nuclear redistribution of cyclin D1 following activation of PKC.
Phenotype of PECs in SSeCKS−/− Mice
We recently generated ssecks gene knockout (SSeCKS−/−) mice,22 and similar to the glomerular phenotype of mice lacking other G1-to-S cell-cycle inhibitors,8 SSeCKS−/− mice showed no gross defects in postnatal, mature glomeruli (Figure 4b) when compared with SSeCKS+/+ mice (Figure 4a) by light microscopy. Examination of electron micrographs of glomeruli from SSeCKS−/− mice uncovered no ultrastructural defects in basement membranes, endothelial cells, podocytes, or within the mesangium (Figure 4c and Supplementary Figure 3). PECs in SSeCKS−/− mice also exhibited no discernable ultrastructural defects, forming tight junctions that appear to be normal (Figure 4d and Supplementary Figure 3).19, 25 This recapitulates the remarkable redundancy of cell-cycle mechanisms to preserve the overall development, structure, and function of the glomerulus in the absence of injurious insults.


Phenotype of glomeruli and parietal epithelial cells (PECs) in SSeCKS−/− mice. (a) Representative periodic acid Schiff (PAS)-stained kidney section from SSeCKS+/+ mice. (b) Representative PAS-stained kidney section from SSeCKS−/− mice showing no glomerular disease by light microscopy. (c) Representative electron micrograph showing no ultrastructural defects in podocytes or glomerular endothelial cells in SSeCKS−/− mice. (d) Representative electron micrograph showing no ultrastructural defects and normal tight junctions (arrows) in PECs in SSeCKS−/− mouse. (e) Representative glomerulus from SSeCKS+/+ mice showing a normal number of paired box gene 8 (PAX8)-positive PECs (arrowheads). (f) Representative glomerulus from SSeCKS−/− mice with same diameter as in panel e showing marked increase in the number of PAX8-positive PECs (arrowheads) compared with SSeCKS+/+ mice. (g) Quantification of the number of PECs per glomerular surface area in cohorts (n=5) of age-matched male SSeCKS+/+ and SSeCKS−/− showing significant basal hyperplasia of PECs in SSeCKS−/− mice. ***P<0.0005. Magnifications: × 200 in panels a and b; × 25 000 in panels c and d; × 400 in panels e and f. SSeCKS, Src-suppressed protein kinase C substrate.
As noted previously, however, SSeCKS serves as a set-point control for the density of cell growth by sequestering and downregulating cyclin D1.13, 14, 15, 16 In SSeCKS−/− mice, the loss of this function contributes to the development of basal hyperplasia of epithelia of the prostate and the epidermis.22, 29 Thus, we quantified the average number of PAX8-positive PECs and WT1-positive podocytes per glomerulus in cohorts of age-matched male SSeCKS+/+ mice (n=5) and male SSeCKS−/− mice (n=5) to determine whether the absence of SSeCKS alters the density of growth of PECs and possibly the replacement of podocytes at steady-state in vivo. Compared with SSeCKS+/+ mice, the number of PAX8-positive PECs per glomerulus in SSeCKS−/− mice was significantly greater, double that normally present in wild-type mice (Figure 4e–g),21 whereas the number of WT1-positive podocytes per glomerulus in SSeCKS+/+ and SSeCKS−/− mice did not significantly differ (17.1±3.2 versus 16.3±2.7, respectively; Supplementary Figure 4). This suggests that PECs develop basal hyperplasia and increased saturation density in the absence of SSeCKS that is not secondary to their inability to replace podocytes. These results indicate that SSeCKS is required to control the density-dependent growth of PECs in vivo, whereas the absence of SSeCKS does not induce developmental or morphological abnormalities in other glomerular cells.
Expression of SSeCKS in Models of Podocytopathies
Studies to trace the proliferation and migration of glomerular epithelial cells in proliferative and cytopenic podocytopathies have shown that PECs are major constituents of the extracapillary lesions that may form following injury to podocytes, possibly representing aberrant glomerular repair by PECs.3, 4, 5 We therefore studied the expression of SSeCKS in archival tissue from models with existing glomerular disease representing a spectrum of podocytopathies,30 specifically the following: Tg26 (a model of collapsing glomerulopathy),20 mice with passive NTN (a model of crescentic glomerulonephritis),24 rats with PAN (a model of minimal change disease),24 rats with PHN (a model of membranous nephropathy),24 and mice with AN (a model of focal segmental glomerulosclerosis).24
Examination of these diseased models suggests that SSeCKS-positive PECs are major constituents of extracapillary glomerular lesions. In the proliferative podocytopathies of Tg26 and NTN, SSeCKS was expressed throughout the crescentic lesions of these models (Figure 5b and c). In contrast, in the non-proliferative podocytopathies of PAN and PHN, which do not readily develop synechial attachments between the capsule and tuft of the glomerulus, the expression of SSeCKS remained limited to PECs on BBM (Figure 5d and e) similar to normal mice (Figure 5a). However, in the cytopenic podocytopathy, AN, which develops segmental sclerosis, SSeCKS was detected in cells migrating over synechial attachments and sclerosing tufts, as well as in cells on BBM (Figure 5f). These results corroborate studies showing that PECs proliferate, migrate, and populate extracapillary lesions,3, 4, 5 and further suggest that SSeCKS may play an important role in determining the pathogenic response of PECs following glomerular injury. Notably, however, these results do not exclude the possibility that SSeCKS may be expressed de novo by diseased podocytes in proliferative and cytopenic podocytopathies.

Expression of Src-suppressed protein kinase C substrate (SSeCKS) in models of podocytopathies. SSeCKS is detected in the glomeruli by immunofluorescence (green) in the following models: (a) normal mice, showing expression of SSeCKS in parietal epithelial cells (PECs) on Bowman's basement membrane (BBM); (b) mice with nephrotoxic nephritis (NTN), showing expression of SSeCKS throughout the proliferative extracapillary lesion; (c) Tg26 mice, showing expression of SSeCKS throughout the proliferative extracapillary lesion; (d) rats with puromycin aminonucleoside nephrosis (PAN), showing expression of SSeCKS in PECs on BBM, similar to normal mice; (e) rats with passive Heymann nephritis (PHN), showing expression of SSeCKS in PECs on BBM, similar to normal mice; and (f) mice with adriamycin nephrosis (AN), showing expression of SSeCKS in PECs on BBM and in cells overlying synechial attachments and sclerosing tufts. Magnifications: × 400 in panels a–f.
Proliferative Injury of PECs in SSeCKS−/− Mice
To address whether the absence of SSeCKS scaffolding function for cyclin D1 alters not only normal density-dependent growth, but also the pathogenic response of PECs following mitogenic injury, we studied the mouse model of passive NTN, in which proliferating PECs are major constituents of extracapillary lesions.31 Challenge of cohorts of age-matched male SSeCKS+/+ mice (n=5) and male SSeCKS−/− mice (n=5) with vehicle induced no pathological glomerular injury (data not shown), and no abnormal or differential proteinuria as measured by urine (albumin/creatinine) on day 14 after challenge (ie, 0.35±0.20 in SSeCKS+/+ mice versus 0.30±0.18 in SSeCKS−/− mice). In contrast, quantification of the percent of total glomeruli with any crescent formation 14 days after challenge with nephrotoxic immunoglobulin showed a significant, fourfold increase in SSeCKS−/− mice when compared with SSeCKS+/+ mice (Figure 6a). Unlike the predominant segmental crescentic lesions in diseased SSeCKS+/+ mice (Figure 6b), many glomeruli in diseased SSeCKS−/− mice showed circumferential, BBM anchorage-independent proliferative lesions with swollen epithelial cells containing hyaline droplets and capillary collapse (Figure 6c). Consistent with this more severe pathology, SSeCKS−/− mice with NTN exhibit 10-fold greater proteinuria than SSeCKS+/+ mice with NTN (Figure 6d), out of proportion to percent crescent formation, suggesting loss of GFB more characteristic of collapsing glomerulopathy than crescentic glomerulonephritis.32, 33

Proliferative injury of parietal epithelial cells (PECs) in nephrotoxic nephritis in SSeCKS−/− mice. (a) Percent of total glomeruli with crescents showing fourfold greater proliferative disease in SSeCKS−/− mice (n=5) compared with SSeCKS+/+ mice (n=5). (b) Representative segmental crescent in diseased glomeruli of SSeCKS+/+ mice. (c) Representative circumferential crescent with capillary collapse in diseased glomeruli of SSeCKS−/− mice. (d) Level of urine (albumin/creatinine) in disease cohorts showing 10-fold greater proteinuria in SSeCKS−/− mice compared with SSeCKS+/+ mice. (e) Quantification of paired box gene 8 (PAX8)-positive nuclei in diseased glomeruli showing a significantly greater number of PECs in diseased SSeCKS−/− mice compared with diseased SSeCKS+/+ mice. (f) Representative immunostaining of PAX8 in glomerular crescents of SSeCKS+/+ mice. (g) Representative immunostaining of PAX8 in glomerular crescents of SSeCKS−/− mice. (h) Quantification of cyclin D1-positive nuclei in diseased glomeruli showing a significantly greater number of positive cells in diseased SSeCKS−/− mice compared with diseased SSeCKS+/+ mice. *P<0.05; **P<0.005; ***P<0.0005. Magnifications: × 400 in panels b, c, f, and g. SSeCKS, Src-suppressed protein kinase C substrate.
We confirmed that proliferating PECs accounted for the more severe extracapillary lesions in SSeCKS−/− mice with NTN. Staining and quantification of the number of PAX8-positive cells in diseased glomeruli showed a significantly higher number of PECs in proliferative lesions present in SSeCKS−/− mice compared with SSeCKS+/+ mice (Figure 6e–g). This was accompanied by a significantly greater number of glomerular cells expressing nuclear cyclin D1 in diseased glomeruli of SSeCKS−/− mice compared with SSeCKS+/+ mice (Figure 6h). Taken together, these results demonstrate that in the absence of SSeCKS scaffolding function, PECs develop a markedly exaggerated and detrimental proliferative response to mitogenic injury in vivo.
DISCUSSION
As progenitors for podocytes,1, 2 mature PECs need robust mechanisms that confer the potential for repeated periods of cell-cycle arrest and re-entry that are distinct from and not active in progeny podocytes.6 These mechanisms are largely unknown. In this study, we show that SSeCKS fulfills the criteria in support of this role in PECs. Parallel to but separate from the expression of other G1-to-S cell-cycle inhibitors, p27 and p57, in embryonic podocytes,8 SSeCKS is induced in embryonic PECs during the later formative stages of glomerulogenesis, and remains constitutively expressed in PECs along the entire BBM in postnatal glomeruli. A stable cytoplasmic pool of cyclin D1 binds SSeCKS in PECs at steady state in vivo and in vitro, and phosphorylation of SSeCKS by aPKC allows for nuclear translocation of this cyclin D1. In the absence of this SSeCKS scaffolding function in vivo, PECs exhibit basal hyperplasia in otherwise normally functioning postnatal glomeruli, in which podocytes and other glomerular cell types show no discernable abnormalities. Taken together, these results suggest that SSeCKS serves as a specific gating checkpoint for cell-cycle arrest of PECs apart from podocytes by sequestering cyclin D1. This distinct role for SSeCKS in PECs fits well with knowledge of the highly divergent, varied tissue-specific functions of member AKAPs.9, 10 These results also highlight the critical role of tumor suppressors to properly maintain niches of tissue progenitor cells.6 However, as with other G1-to-S cell-cycle inhibitors,8 absence of SSeCKS does not appear to be sufficient alone to induce clinical glomerular disease.
Nevertheless, our studies of NTN in SSeCKS−/− mice suggest that lack of the ssecks/akap12 gene confers susceptibility for and increased severity of proliferative podocytopathies by allowing for exaggerated proliferation of PECs. In both crescentic glomerulonephritis and collapsing glomerulopathy, cells derived from PECs are now known to be major constituents of these extracapillary lesions, possibly representing an aberrant hyperplastic repair response by PECs.3, 4, 5 We hypothesized earlier that patterns of aberrant repair in the proliferative podocytopathies lie on a spectrum of inflammatory injury,32, 33, 34 recently identifying the TNF-α–TNFR2 axis as one possible causative pathway.27, 28 Intriguingly, TNF-α is one of a host of mitogenic activators of PKC,26 inducing PKC phosphorylation of SSeCKS in PECs. This phosphorylation disrupts SSeCKS ability to bind and sequester cyclin D112, 13, 14 in regulating the proliferative response of PECs, a state mimicked by the lack of SSeCKS in SSeCKS−/− mice. Notably, however, unlike the loss of SSeCKS expression and scaffolding function that occurs in malignancies from genetic deletion of ssecks/akap12,9 our staining of podocytopathy models containing ssecks/akap12 shows that SSeCKS expression remains in PECs forming non-malignant extracapillary lesions. Thus, the cyclic, temporal loss of SSeCKS scaffolding function in proliferating PECs in diseased glomeruli likely occurs by its rapid phosphorylation and de-phosphorylation, as known for AKAPs,10 which we cannot detect in vivo with our current reagents. In any respect, our results extend that of others suggesting that diseased PECs may play an important role in determining the morphology and clinical outcome following glomerular injury,3, 4, 5 with SSeCKS being one newly identified determinant of the cellular response by PECs.
In additional to regulating cyclin D1, SSeCKS may control other molecular pathways in PECs of importance to their function as progenitors for podocytes, as coordinating multiple signaling pathways is the defining feature of AKAPs such as SSeCKS.9, 10 For example, by scaffolding and attenuating PKCα/δ, SSeCKS suppresses pathways that induce senescence,35 a ‘care-taking’ role of some tumor suppressors to maintain niches of tissue progenitors.6 This would be a rationale function for SSeCKS in PECs, which theoretically must continually self-renew without aging to replace podocytes throughout life. In addition, little is known of the intrinsic molecular events that govern the phenotypic transformation of PECs to podocytes, for example, from constitutively suppressing to constitutively expressing vascular endothelial growth factor (VEGF) or altering the secretion of extracellular matrices for BBM to extracellular matrices for the glomerular basement membrane. Interestingly, SSeCKS suppresses the expression of VEGF,36, 37 accounting in part for its inhibition of metastatic growth, raising the possibility for a similar role in PECs, whereas any role for SSeCKS in the production of extracellular matrices by PECs is unknown.38 These examples highlight additional avenues for future research on the unique function of SSeCKS in PECs apart from podocytes.
References
Romagnani P . Toward the identification of a ‘renopoietic system’? Stem Cells 2009;27:2247–2253.
Lasagni L, Romagnani P . Glomerular epithelial stem cells: the good, the bad, and the ugly. J Am Soc Nephrol 2010;21:1612–1619.
Appel D, Kershaw DB, Smeets B, et al. Recruitment of podocytes from glomerular parietal epithelial cells. J Am Soc Nephrol 2009;20:333–343.
Smeets B, Uhlig S, Fuss A, et al. Tracing the origin of glomerular extracapillary lesions from parietal epithelial cells. J Am Soc Nephrol 2009;20:2604–2615.
Smeets B, Angelotti ML, Rizzo P, et al. Renal progenitor cells contribute to hyperplastic lesions of podocytopathies and crescentic glomerulonephritis. J Am Soc Nephrol 2009;20:2593–2603.
He S, Nakada D, Morrison SJ . Mechanisms of stem cell self-renewal. Annu Rev Cell Dev Biol 2009;25:377–406.
Petermann A, Hiromura K, Pippin J, et al. Differential expression of d-type cyclins in podocytes in vitro and in vivo. Am J Pathol 2004;164:1417–1424.
Marshall CB, Shankland SJ . Cell cycle regulatory proteins in podocyte health and disease. Nephron Exp Nephrol 2007;106:e51–e59.
Gelman IH . The role of SSeCKS/gravin/AKAP12 scaffolding proteins in the spatiotemporal control of signaling pathways in oncogenesis and development. Front Biosci 2002;7:d1782–d1797.
Carnegie GK, Means CK, Scott JD . A-kinase anchoring proteins: from protein complexes to physiology and disease. IUBMB Life 2009;61:394–406.
Lin X, Nelson PJ, Frankfort B, et al. Isolation and characterization of a novel mitogenic regulatory gene, 322, which is transcriptionally suppressed in cells transformed by src and ras. Mol Cell Biol 1995;15:2754–2762.
Lin X, Tombler E, Nelson PJ, et al. A novel src- and ras-suppressed protein kinase C substrate associated with cytoskeletal architecture. J Biol Chem 1996;271:28430–28438.
Lin X, Nelson P, Gelman IH . SSeCKS, a major protein kinase C substrate with tumor suppressor activity, regulates G1 → S progression by controlling the expression and cellular compartmentalization of cyclin D. Mol Cell Biol 2000;20:7259–7272.
Lin X, Gelman IH . Calmodulin and cyclin D anchoring sites on the Src-suppressed C kinase substrate, SSeCKS. Biochem Biophys Res Commun 2002;290:1368–1375.
Nelson PJ, Daniel TO . Emerging targets: molecular mechanisms of cell contact-mediated growth control. Kidney Int 2002;61 (Suppl):S99–105.
Nelson PJ, Gelman IH . Cell-cycle regulated expression and serine phosphorylation of the myristylated protein kinase C substrate, SSeCKS: correlation with culture confluency, cell cycle phase and serum response. Mol Cell Biochem 1997;175:233–241.
Griner EM, Kazanietz MG . Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer 2007;7:281–294.
Nelson PJ, Moissoglu K, Vargas Jr J, et al. Involvement of the protein kinase C substrate, SSeCKS, in the actin-based stellate morphology of mesangial cells. J Cell Sci 1999;112 (Part 3):361–370.
Ohse T, Chang AM, Pippin JW, et al. A new function for parietal epithelial cells: a second glomerular barrier. Am J Physiol Renal Physiol 2009;297:F1566–F1574.
Barisoni L, Madaio MP, Eraso M, et al. The kd/kd mouse is a model of collapsing glomerulopathy. J Am Soc Nephrol 2005;16:2847–2851.
Ohse T, Vaughan MR, Kopp JB, et al. De novo expression of podocyte proteins in parietal epithelial cells during experimental glomerular disease. Am J Physiol Renal Physiol 2010;298:F702–F711.
Akakura S, Huang C, Nelson PJ, et al. Loss of the SSeCKS/Gravin/AKAP12 gene results in prostatic hyperplasia. Cancer Res 2008;68:5096–5103.
Ohse T, Pippin JW, Vaughan MR, et al. Establishment of conditionally immortalized mouse glomerular parietal epithelial cells in culture. J Am Soc Nephrol 2008;19:1879–1890.
Pippin JW, Brinkkoetter PT, Cormack-Aboud FC, et al. Inducible rodent models of acquired podocyte diseases. Am J Physiol Renal Physiol 2009;296:F213–F229.
Ohse T, Pippin JW, Chang AM, et al. The enigmatic parietal epithelial cell is finally getting noticed: a review. Kidney Int 2009;76:1225–1238.
Schutze S, Machleidt T, Kronke M . The role of diacylglycerol and ceramide in tumor necrosis factor and interleukin-1 signal transduction. J Leukocyte Biol 1994;56:533–541.
Vielhauer V, Stavrakis G, Mayadas TN . Renal cell-expressed TNF receptor 2, not receptor 1, is essential for the development of glomerulonephritis. J Clin Invest 2005;115:1199–1209.
Bruggeman LA, Drawz PE, Kahoud N, et al. TNFR2 interposes the proliferative and NF-kappaB-mediated inflammatory response by podocytes to TNF-alpha. Lab Invest 2011;91:413–425.
Akakura S, Bouchard R, Bshara W, et al. Carcinogen-induced squamous papillomas and oncogenic progression in the absence of the SSeCKS/AKAP12 metastasis suppressor correlates with FAK upregulation. Int J Cancer 2011;129:2025–2031.
Wiggins RC . The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int 2007;71:1205–1214.
Ophascharoensuk V, Pippin JW, Gordon KL, et al. Role of intrinsic renal cells versus infiltrating cells in glomerular crescent formation. Kidney Int 1998;54:416–425.
Albaqumi M, Soos TJ, Barisoni L, et al. Collapsing glomerulopathy. J Am Soc Nephrol 2006;17:2854–2863.
Barisoni L, Nelson PJ . Collapsing glomerulopathy: an inflammatory podocytopathy? Curr Opin Nephrol Hypertens 2007;16:192–195.
Bruggeman LA, Nelson PJ . Controversies in the pathogenesis of HIV-associated renal diseases. Nat Rev 2009;5:574–581.
Akakura S, Nochajski P, Gao L, et al. Rb-dependent cellular senescence, multinucleation and susceptibility to oncogenic transformation through PKC scaffolding by SSeCKS/AKAP12. Cell Cycle 2010;9:4656–4665.
Lee SW, Kim WJ, Choi YK, et al. SSeCKS regulates angiogenesis and tight junction formation in blood–brain barrier. Nat Med 2003;9:900–906.
Su B, Zheng Q, Vaughan MM, et al. SSeCKS metastasis-suppressing activity in MatLyLu prostate cancer cells correlates with vascular endothelial growth factor inhibition. Cancer Res 2006;66:5599–5607.
Smeets B, Kuppe C, Sicking E, et al. Parietal epithelial cells participate in the formation of sclerotic lesions in focal segmental glomerulosclerosis. J Am Soc Nephrol 2011;22:1262–1274.
Acknowledgements
We thank Lingqiu Gao for assistance with studies using anti-phospho-SSeCKS ser283 antibody and John Scott for helpful discussions. This work was supported by the National Institutes of Health Grants DK83375 (to PJN), DK83375S1 (to PJN), DK56799 (to SJS), DK81835 (to SJS), and DK76126 (to CEA). PJN is a member of the Nephrotic Syndrome Study Network funded by DK83912.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
Glomerular parietal epithelial cells (PECs) are progenitors for podocytes but the mechanisms that allow for repeated periods of cell-cycle arrest and re-entry of PECs after glomerulogenesis are unknown. The multivalent scaffolding PKC substrate, SSeCKS, regulates the activity of cyclin D1 in PECs, serving as a gating checkpoint for cell-cycle arrest of PECs but not podocytes.
Supplementary information
Rights and permissions
About this article
Cite this article
Burnworth, B., Pippin, J., Karna, P. et al. SSeCKS sequesters cyclin D1 in glomerular parietal epithelial cells and influences proliferative injury in the glomerulus. Lab Invest 92, 499–510 (2012). https://doi.org/10.1038/labinvest.2011.199
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2011.199
Keywords
This article is cited by
-
Parietal epithelial cell dysfunction in crescentic glomerulonephritis
Cell and Tissue Research (2021)
-
Inhibition of mTOR delayed but could not prevent experimental collapsing focal segmental glomerulosclerosis
Scientific Reports (2020)
-
β-catenin regulates the formation of multiple nephron segments in the mouse kidney
Scientific Reports (2019)
-
SSeCKS promoted lipopolysaccharide-sensitized astrocytes migration via increasing β-1,4-galactosyltransferase-I activity
Neurochemical Research (2019)
-
Aldosterone Impairs Mitochondrial Function in Human Cardiac Fibroblasts via A-Kinase Anchor Protein 12
Scientific Reports (2018)
