
Abstract
We have previously demonstrated that calmodulin (CaM) binds directly to c-FLIPL in a Ca2+-dependent manner. Deletion of the CaM-binding region (amino acid 197–213) results in reduced CaM binding, and increased Fas-mediated apoptosis and decreased tumorigenesis of cholangiocarcinoma cells. The present studies were designed to identify the precise amino acids between 197 and 213 that are responsible for CaM/FLIP binding, and their roles in mediating the anti-apoptotic function of c-FLIPL. Sequence analysis of the CaM-binding region at 197–213 predicted three unique positively charged residues at 204, 207 and 209, which might be responsible for the CaM/FLIP binding. A point mutation at H204 of c-FLIPL was found to markedly reduce CaM binding, whereas point mutation at R207 or K209 did not affect c-FLIPL binding to CaM. Decreased CaM/FLIP binding was confirmed in cholangiocarcinoma cells overexpressing the H204 c-FLIPL mutant. Reduced CaM binding by the H204 mutant resulted in increased sensitivity to Fas-mediated apoptosis and inhibited tumor growth in mice compared with wild-type c-FLIPL. Death-inducing signaling complex (DISC) analysis showed that the reduced CaM binding to H204 mutant resulted in less c-FLIPL recruited into the DISC. Concurrently, increased caspase 8 was recruited to the DISC, which resulted in increased cleavage and activation of caspase 8, activation of downstream caspase 3 and increased apoptosis. Therefore, these results demonstrate that the H204 residue is responsible for c-FLIPL binding to CaM, which mediates the anti-apoptotic function of c-FLIPL, most likely through affecting recruitment of caspase 8 into the DISC and thus caspase 8 activation. These studies further characterized CaM/FLIP interaction and its function in regulating Fas-mediated apoptosis and tumorigenesis, which may provide new therapeutic targets for cancer therapy.
Similar content being viewed by others


Main
Apoptosis is an important process in a wide variety of different biological systems, including normal cell turnover, development of the immune system, embryonic development, metamorphosis, hormone-dependent atrophy and also in chemical-induced cell death.1 Stimulation of apoptosis has been promoted as a potential therapy for many cancers, including cholangiocarcinoma. Results from our group and others have suggested that regulation of Fas-mediated apoptosis is a promising therapeutic avenue for cholangiocarcinoma.2, 3, 4, 5, 6, 7, 8, 9
The Fas death receptor is a member of the tumor necrosis factor (TNF) receptor superfamily. In response to the binding of its ligand (FasL) or agonistic antibody, Fas undergoes oligomerization and its death domain (DD) binds to the adaptor molecule Fas-associated DD (FADD) through a homotypic interaction.10 Then, FADD recruits and aggregates caspase 8 and/or cellular FLICE-like inhibitory protein (c-FLIP) to form the death-inducing signaling complex (DISC). Death or survival signals transmitted through the DISC are determined by the relative expression of caspase 8 or its homolog with no caspase activity, c-FLIP, recruited into the DISC.11, 12, 13, 14 Two cellular isoforms of FLIP have been identified, c-FLIPL (55 kDa) and c-FLIPs (26 kDa). c-FLIPs contain only the tandem death effector domains (DEDs). c-FLIPL contains not only the tandem DEDs, but also a protease-like domain, homologous to caspase 8, in which several amino acids important for protease activity are mutated, including the cysteine at the active site.15 c-FLIPL has generally been thought to be an anti-apoptotic protein, however, some evidence suggests that it can also improve Fas-mediated apoptosis by forming a heterodimer with caspase 8, which helps achieve the initial cleavage step of procaspase 8.16, 17, 18 With stable expression of caspase 8, c-FLIPL is believed to inhibit caspase 8 activation by interfering with the recruitment and/or activation of caspase 8 in the DISC.16, 17, 19 However, the precise mechanism for the anti-apoptotic function of c-FLIPL in Fas-mediated apoptosis is not clearly understood.
Our previous studies have demonstrated that calmodulin (CaM) antagonists induce apoptosis through a Fas-related mechanism in a cholangiocarcinoma tumor model.2, 3, 5, 9 CaM is a small (17 kDa), acidic, dumbbell-shaped protein that binds Ca2+ through EF-hand motifs.20 Its important role in apoptosis has been reported in several studies.21, 22, 23 We have shown that CaM binds directly to Fas.24 Further studies on the interaction of CaM with other proteins in the Fas-induced DISC showed that CaM also directly binds to c-FLIPL in a Ca2+-dependent manner, but not FADD or caspase 8.25 The CaM-binding region in c-FLIPL was identified between amino acids 197 and 213. Deletion of this region causes reduced binding between CaM and c-FLIPL. Overexpression of c-FLIPL lacking this region results in increased Fas-mediated apoptosis in cholangiocarcinoma cells and decreased tumorigenesis compared with wild-type (WT) c-FLIPL-overexpressing cells.2, 25 These findings indicate that CaM-c-FLIPL binding is important for the anti-apoptotic function of c-FLIPL in the Fas pathway.
Sequence comparison of the CaM-binding region at 197–213 on c-FLIPL with those of c-FLIPS and caspase 8 predicted three unique positively charged residues at 204, 207 and 209, which might be responsible for the CaM/FLIP binding. We found that point mutation at H204 significantly reduced the binding to CaM, whereas point mutation at R207 or K209 showed no effect. Reduced CaM binding by the c-FLIPL H204 mutant in cholangiocarcinoma cells increased their sensitivity to Fas-mediated apoptosis and decreased their tumorigenic potential in a nude mouse model compared with WT c-FLIPL. DISC analysis, further demonstrated that reduced CaM binding to c-FLIPL resulted in less c-FLIPL and increased the caspase 8 recruited into the DISC, which resulted in increased activation of caspase 8, and thus activation of downstream signals and increased apoptosis. Our findings provide new evidence and mechanistic molecular insight to support the concept that CaM-c-FLIPL binding modulates the anti-apoptotic function of c-FLIPL in Fas-mediated apoptosis and consequently the tumorigenic potential of cancer cells.
MATERIALS AND METHODS
Cell Culture, Antibodies and Reagents
The cholangiocarcinoma cell line, SK-ChA-1, is one of three human cell lines from adenocarcinomas of the extrahepatic biliary tract established by Dr A Knuth (Ludwig Institute for Cancer Research, London, UK).26 Cells were grown in RPMI 1640 (Invitrogen) supplemented with penicillin (5 units/ml), streptomycin (5 μg/ml) and 10% heat-inactivated fetal bovine serum. Cholangiocarcinoma cell line, Mz-ChA-1, was kindly provided by Dr Gregory J Gores (Mayo Clinic, Rochester, MN, USA) and maintained as previously described.2, 27 The FLIP antibody, NF-6, was obtained from Alexis Biochemicals. The caspase 3 antibody was from StressGen and antibodies to caspase 8 and FADD were purchased from BD Bioscience. Fas-activating antibody, clone CH-11, was from Upstate Biotechnology. The monoclonal antibody to CaM was developed as previously described.28 Antibodies to Fas and GAPDH were purchased from Santa Cruz Biotechnology.
Expression and Purification of His-SUMO Fusion Proteins
The human c-FLIPL cDNA was subcloned into the pET28a vector with His-SUMO provided by Dr J Ma (Department of Biochemistry and Molecular Genetics, UAB, Birmingham, AL) under Bgl II and Xho I sites. Point mutations H204, R207 and K209 in c-FLIPL were generated with a QuikChange site-directed mutagenesis kit (Stratagene) and confirmed by sequencing. The recombinant c-FLIPL proteins were expressed in E. coli BL21-Gold (DE3) (Stratagene). After induction with 0.1 mM IPTG, the cells were allowed to grow at 30°C overnight. Collected cells were lysed by adding lysosome and sonication. Proteins were purified by Ni-NTA Superflow Columns (Qiagen).
Protein Pulldown
Protein pulldown with CaM–Sepharose 4B (Amersham Biosciences) was performed as described previously.24, 25 Briefly, 20 μg of purified fusion protein or 300 μg of extracted protein from cholangiocarcinoma cells overexpressing WT c-FLIPL or the H204 mutant was incubated with CaM–Sepharose 4B in the lysis buffer (20 mM Tris-HCl, 150 mM NaCl, 10% glycerol, 1% Triton X-100 and proteinase inhibitor mix, pH 7.5) with 2 mM CaCl2 or 2 mM EGTA overnight at 4°C. The beads were washed four times with lysis buffer and proteins were identified by western blot analysis.
Western Blot Analysis
Protein extracts from cells were prepared as described previously.2, 3 Concentrations of protein were determined with a BCA protein assay kit (Thermo Scientific). Proteins were separated by SDS–PAGE and transferred to Immobilon P membranes (Millipore) as described previously.2, 3 Membranes were blocked in 5% non-fat milk and incubated with primary antibodies overnight at 4°C. Horseradish peroxidase-conjugated secondary antibodies in the blocking buffer were incubated for 1 h at room temperature. Signals were detected using Immobilon Western chemiluminescent horseradish peroxidase substrate detection kit (Millipore). Bands were quantified by ImageQuant version 5.1 (GE Healthcare Lifesciences).
Generation of Cholangiocarcinoma Cells Stably Overexpressing WT c-FLIPL and H204 Mutant
Stable overexpression of c-FLIPL and the H204 c-FLIPL mutant in cholangiocarcinoma cells, SK-ChA-1, was accomplished using methods described previously.29, 30 The human WT c-FLIPL and H204 mutant cDNA were subcloned into the lentiviral vector under Bgl II and Xho I sites. Each construct was packed into lentivirus-like particles pseudotyped with the vesicular stomatitis virus glycoprotein as we previously described.31 Transduction was performed by incubating cholangiocarcinoma cells with recombinant lentivirus, and stably transduced cells were selected with puromycin (2 μg/ml).
Transient Transfection Assays
The human WT c-FLIPL and H204 mutant cDNA were subcloned into the pcDNA3.1. vector under Bgl II and Xho I sites and confirmed by sequencing. Cholangiocarcinoma cells, SK-ChA-1 or Mz-ChA-1, were transfected with these constructs at a 1:3 ratio using Fugene 6 (Roche Applied Science). Twenty-four hours after transfection, cells were harvested or treated with Fas-activating antibody, CH-11, for apoptosis assay.
Assessment of Apoptosis
Apoptosis was induced with Fas-activating antibody, CH-11, as described previously.4 Briefly, cholangiocarcinoma cells were exposed to 500 ng/ml of CH-11 antibody for 24 h. Apoptosis was determined by Annexin V-FITC and propidium iodide staining (BD biosciences) and analyzed by flow cytometry (BD biosciences).
Analysis of Fas-Mediated DISC
Immunoprecipitation for DISC analysis was performed as previously described.32 Cholangiocarcinoma cells (5 × 107), SK-ChA-1, were incubated with or without 1 μg/ml Fas-activating antibody (CH-11) for 30 or 60 min at 37°C and then washed with PBS and lysed in lysis buffer (20 mM Tris-HCl, 150 mM NaCl, 10% glycerol, 1% Triton X-100 and proteinase inhibitor mix, pH 7.5) for 30 min on ice. In control cells, Fas-activating antibody (CH-11) was added to cell lysates at a final concentration of 1 μg/ml to immunoprecipitate non-stimulated Fas receptors. After centrifugation at 13 000 g for 15 min at 4°C, the supernatant was incubated with 20 μl of goat anti-mouse IgM agarose (Sigma) overnight at 4°C and analyzed by western blot analysis.
Mouse Xenograft Model
The animal protocol was approved by the Institutional Animal Care and Use Committee at the University of Alabama at Birmingham, Birmingham, AL, USA. Male athymic nu/nu mice (4 weeks, NCI-Frederick) were used for tumor inoculation. Briefly, cholangiocarcinoma cells, SK-ChA-1, stably overexpressing WT c-FLIPL or the H204 mutant (2 × 106 cells in 200 μl PBS/site) were inoculated subcutaneously into the flank area of mice. Tumor size and body weight were measured every 3 days and volumes were determined using the formula volume=length × width2/2.
TUNEL Staining
At the end of the animal experiment, tumors were removed, fixed in 4% paraformaldehyde and embedded in paraffin. Consecutive tumor sections (8 μm) from each group were used for histological and immunohistochemial staining. To assess apoptotic cells in tumor tissues, TUNEL staining (DeadEnd Fluorometric TUNEL System; Promega) was performed following the manufacturer's protocol. For quantitative analysis, cell numbers were counted under a microscope ( × 400). Four fields in each slide were counted and the percentage of apoptotic cells was determined.
Statistical Analysis
Results are expressed as means±s.e. Differences between two groups were identified with Student's t-test. Significance was defined as P<0.05.
RESULTS
c-FLIPL H204 Mutant has Reduced Binding to CaM In Vitro
Previous studies from our group demonstrated the CaM-binding region in c-FLIPL is amino acids 197–213.25 To identify the precise amino acids between 197 and 213 that are responsible for CaM/FLIP binding, we further compared the 197–213 regions of c-FLIPL, c-FLIPS and caspase 8. As shown in Figure 1a, c-FLIPL has more positive charges, which was largely due to three amino acids (H204, R207 and K209). Since c-FLIPS and caspase 8 do not bind to CaM, these three amino acids might be essential for the interaction between c-FLIPL and CaM. To test this hypothesis, H204, R207 and K209 were mutated to alanine separately with a QuikChange site-directed mutagenesis kit as mentioned in the Materials and Methods. These c-FLIPL mutations and WT were cloned into a His-SUMO vector and proteins were expressed in E. coli using IPTG induction and purified with Ni-NTA Superflow Columns. Binding to CaM was analyzed with a pulldown assay using CaM-Sepharose 4B. As shown in Figures 1b and d, only the H204 mutant showed reduced binding to CaM, whereas R207 and K209 had no effect. Furthermore Ca2+ did not significantly affect the H204 mutant binding to CaM in contrast to WT c-FLIPL, in which CaM/FLIP binding was largely Ca2+-dependent, being decreased substantially with the presence of EGTA (Figures 1c and e).

c-FLIPL H204 mutant has reduced binding to CaM in vitro. (a) Sequence comparison of the 197–213 regions from c-FLIPL, c-FLIPS and caspase 8. Amino acids with positive (red, underline) or negative (blue, italic) charge are indicated. Protein pulldown assays with CaM-Sepharose 4B were performed using (b, d) various His-SUMO-FLIPL mutations or (c, e) His-SUMO-FLIPL WT and H204 mutant in the presence of Ca2+ or EGTA. Proteins binding to CaM-Sepharose 4B were determined by western blot analysis. Results shown are means±s.e. (n=3). ##P<0.01 for comparison with WT c-FLIPL.
c-FLIPL H204 Mutant has Reduced Binding to CaM in Cholangiocarcinoma Cells and Sensitizes the Cells to Fas-Induced Apoptosis
To determine whether the reduced c-FLIPL binding to CaM by the H204 mutant also exists in cells, we stably overexpressed WT c-FLIPL and H204 mutant in cholangiocarcinoma cells, SK-ChA-1, using the lentiviral expression vectors (Figure 2a). Whole cell lysates from these stable overexpression cells were used for protein pulldown assays with CaM-Sepharose 4B. Consistent with the in vitro data, the H204 mutant reduces c-FLIPL binding to CaM compared with WT c-FLIPL (Figures 2b and c). The CaM-c-FLIPL interaction has been described as an important modulator of Fas-mediated apoptosis, which is an important pathway for apoptotic cell death in cholangiocarcinoma cells.16, 25 To determine the effect of the reduced CaM-c-FLIPL interaction by H204 on Fas-mediated apoptosis, cholangiocarcinoma cells overexpressing LacZ, WT c-FLIPL or H204 mutant were treated with CH-11, the Fas-activating antibody, for 24 h. Apoptosis was determined by Annexin V/propidium iodide staining. As shown in Figure 2d, overexpression of WT c-FLIPL inhibited Fas-mediated apoptosis compared with control. The H204 mutant-overexpressing cells were partially resistant to Fas-mediated apoptosis as compared with controls, however, their sensitivity to Fas-mediated apoptosis was significantly higher than WT c-FLIPL-overexpressing cells (Figure 2d). Thus the c-FLIPL H204 mutant partially rescues the inhibition of WT c-FLIPL on Fas-mediated apoptosis most likely because of the reduced binding with CaM.25 Neither WT c-FLIPL nor the H204 mutant affected the levels of basal apoptosis (Figure 2d).

c-FLIPL H204 mutant has reduced binding to CaM in cholangiocarcinoma cells and sensitizes the cells to Fas-induced apoptosis. (a) Western blot analysis for c-FLIPL expression using cell lysates from cholangiocarcinoma cells, SK-ChA-1, stably overexpressing LacZ, WT c-FLIPL or H204 mutant. (b, c) Protein pulldown assays with CaM-Sepharose 4B were performed using whole cell lysates from SK-ChA-1 cells stably overexpressing LacZ, WT c-FLIPL or H204 mutant. The proteins pulled down were immunoblotted for c-FLIPL. (d) Apoptosis was determined by Annexin V/propidium iodide staining in SK-ChA-1 cells stably overexpressing LacZ, WT c-FLIPL or H204 mutant with or without CH-11, the Fas-activating antibody, treatment. Results shown are means±s.e. (n=3). **P<0.01 for comparison with controls. ##P<0.01 and #P<0.05 for comparison of the H204 mutant with WT c-FLIPL.
c-FLIPL H204 Mutant Reduces the Inhibitory effect of WT c-FLIPL on Fas-Induced Caspase Activation
To elucidate the mechanisms underlying the effects of WT c-FLIPL and the H204 mutant on Fas-mediated apoptosis, we determined the expression/activation of caspases in CH-11-treated cholangiocarcinoma cells (SK-ChA-1) overexpressing LacZ, WT c-FLIPL or the H204 mutant. As shown in Figures 3a and c, activation of caspase 3, a downstream caspase, was inhibited by WT c-FLIPL compared with control. Although compared with control the H204 mutant also inhibited activation of caspase 3, activated caspase 3 was higher than that in WT c-FLIPL cells (Figures 3a and c). These results are consistent with the apoptosis data (Figure 2d). c-FLIPL has been thought to act primarily as an anti-apoptotic protein that interferes with activation of caspase 8 as well as recruitment of caspase 8 into the DISC.17, 25 Therefore expression/activation of caspase 8 was determined by western blot analysis. As expected, both overexpression of WT c-FLIPL and the H204 mutant significantly blocked caspase 8 activation by CH-11 compared with controls (Figures 3a and b). However, the H204 mutant partially rescued the inhibition by WT c-FLIPL on caspase 8 activation, suggesting that the CaM binding to c-FLIPL modulates Fas-mediated apoptosis through activating caspase 8.

c-FLIPL H204 mutant reduces the inhibitory effect of WT c-FLIPL on Fas-induced caspase activation. Cholangiocarcinoma cells, SK-ChA-1, stably overexpressing LacZ, WT c-FLIPL or H204 mutant were treated with CH-11 for 24 h. (a) Different apoptosis-related proteins as indicated were detected by western blot analysis. Cleavage of caspase 8 (b) and caspase 3 (c) are quantitated. Results shown are means±s.e. (n=3). **P<0.01 for comparison with controls and #P<0.05 for comparison of the H204 mutant with WT c-FLIPL.
Effects of c-FLIPL H204 Mutant on Fas-Mediated DISC Formation
To determine the mechanism that enables the CaM-c-FLIPL interaction to modulate the sensitivity of cells to Fas-mediated apoptosis, Fas-mediated DISC analysis was performed. Cholangiocarcinoma cells, SK-ChA-1, overexpressing LacZ, WT c-FLIPL or H204 mutant were exposed to CH-11 for 0, 30 and 60 min. The recruitment of c-FLIPL, caspase 8, Fas, CaM and FADD into the DISC in response to CH-11 was analyzed. More WT c-FLIPL and H204 mutant were recruited into the DISC than in control cells because they were overexpressed (Figure 4). Compared with WT c-FLIPL, less c-FLIPL H204 mutant was in the DISC, which supports our previous hypothesis that disrupted CaM-c-FLIPL binding causes a conformational change in c-FLIPL that affects its recruitment into the DISC.25 Recruitment of caspase 8 into DISC was inhibited by overexpression of both WT c-FLIPL and the H204 mutant apparently because of its competition with caspase 8 for FADD binding.10, 33, 34 Activated caspase 8 (p43/41) appeared at 30 min in c-FLIPL H204 mutant cells, which was earlier than 60 min in WT c-FLIPL cells (Figure 4). The amount of CaM in the DISC was decreased in c-FLIPL H204 mutant cells, which confirmed that reduced binding with CaM does exist both in vitro and in vivo (Figures 1 and 2).

Effects of c-FLIPL H204 mutant on Fas-mediated DISC formation. Cholangiocarcinoma cells, SK-ChA-1, stably overexpressing LacZ, WT c-FLIPL or H204 mutant were exposed to CH-11 for 0, 30 and 60 min. Western blot analyses were performed with extracted proteins to determine the expression of DISC proteins including c-FLIPL, caspase 8, Fas, CaM and FADD in response to CH-11 (left panel inputs). Immunoprecipitation for DISC analysis was performed to determine the recruitment of c-FLIPL, caspase 8, Fas, CaM and FADD into DISC in response to CH-11 (right panel-DISC-IP).
Effects of c-FLIPL H204 Mutant on Cholangiocarcinoma Tumorigenesis in Mice
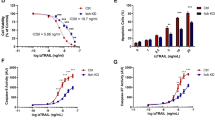
Increased sensitivity to Fas-mediated apoptosis by reduced binding between the H204 mutant and CaM (Figure 2d) suggests that the effects of WT c-FLIPL and the H204 mutant on tumorigenesis may be different. To test this hypothesis, tumorigenesis of WT- and H204 mutant-overexpressing cholangiocarcinoma cells (SK-ChA-1) was characterized in a nude mice xenograft model. Cholangiocarcinoma cells stably overexpressing WT c-FLIPL or the H204 mutant were inoculated subcutaneously into the flank area of 4-week-old male athymic nude mice. Tumor volumes were measured every 3 days. As shown in Figure 5a, H204 mutant overexpressing cells formed significantly smaller tumors than WT c-FLIPL cells from day 9 through day 15. Furthermore, TUNEL staining showed the increased apoptotic cells in H204 mutant tumors compared with WT c-FLIPL tumors (Figures 5b and c). These findings support the concept that the interaction between CaM and FLIP is important in cholangiocarcinoma pathogenesis.2

Effects of c-FLIPL H204 mutant on cholangiocarcinoma tumorigenesis in mice. Cholangiocarcinoma cells, SK-ChA-1, stably overexpressing WT c-FLIPL or H204 mutant were inoculated subcutaneously into the flank area of 4-week-old male athymic nude mice (five mice per group). (a) Tumor volumes were measured every 3 days for 15 days and determined using the formula volume=length × width2/2. (b) At the end of the tumorigenesis experiment, tumors were processed as described in Materials and methods. Consecutive sections of tumors from each mouse were analyzed by H&E staining and TUNEL staining. Representative images of each group are shown. For quantitative analysis, cell numbers were counted and the percentage of positive cells was determined. (c) Percentage of apoptotic cells determined by TUNEL staining. Results shown are means±s.e. (n=10 for tumors and n=12 for TUNEL staining). ##P<0.01 and #P<0.05 for comparison of the H204 mutant with WT c-FLIPL.
DISCUSSION
CaM interacts with a variety of proteins such as calcineurin, CaM kinases, myosin light-chain kinase, nitric oxide synthase and neuromodulin35 and it affects numerous signaling pathways involved in such diverse processes such as inflammation, memory, muscle contraction, the immune response and ion channel functioning.36 Previously, we have shown that CaM binds to two key proteins (Fas and c-FLIPL, but not FADD or caspase 8) in the cell death pathway suggesting a novel role of CaM in Fas-mediated signaling.24, 25 CaM binds to c-FLIPL directly in a Ca2+-dependent manner.25 A CaM-binding region was identified in amino acids 197–213 of c-FLIPL and deletion of this region causes reduced binding between CaM and c-FLIPL.25 Furthermore, deletion of the CaM-binding region results in increased Fas-mediated apoptosis suggesting that CaM-c-FLIPL binding is important for the anti-apoptotic function of c-FLIPL in the Fas pathway.25 To identify the precise amino acids between 197 and 213 that are responsible for CaM/FLIP binding, and their roles in mediating the anti-apoptotic function of c-FLIPL, sequence comparison of this binding region among c-FLIPL, c-FLIPS and caspase 8 predicted three positively charged residues at 204, 207 and 209, which might be responsible for the CaM/FLIP binding (Figure 1a). Considering these findings, it was reasonable to determine whether one of these three amino acid mutants affects c-FLIPL binding to CaM. We found that only the H204 mutant showed reduced binding to CaM, which was confirmed by both using purified proteins and intact cells (Figures 1 and 2). Furthermore, the H204 mutant increased the sensitivity of cells to Fas-mediated apoptosis (Figure 2d) and showed increased apoptosis and decreased tumorigenesis in mice compared with WT c-FLIPL (Figure 5), which provided new evidence that the anti-apoptotic effect of c-FLIPL requires CaM binding. The same mechanism was confirmed by using transiently transfected cells and in an additional cholangiocarcinoma cell line, Mz-ChA-1 (Supplementary Figures 1 and 2).
The function of c-FLIPL in apoptotic signaling is considered to be primarily an anti-apoptotic protein although some evidence indicates that it can enhance Fas-mediated apoptosis when expressed at much lower and higher physiological levels.16, 17, 18 During the early stages of death receptor signaling, the key regulatory protein, c-FLIPL is recruited to the DISC, where it competes with procaspase 8 and 10 binding to FADD and modulates activation of procaspase 8 and 10.17, 37, 38 To elucidate how CaM-c-FLIPL binding modulates the anti-apoptotic function of c-FLIPL, we determined the effects on expression/activation of apoptosis-related proteins. Reduced binding between c-FLIPL and CaM increased caspase 8 activation, which in turn leads to activated caspase 3 and increased apoptosis (Figure 3). Thus CaM-c-FLIPL binding modulates Fas-mediated apoptosis at least in part by modulating activation of caspase 8.
Inhibition of caspase 8 activation by c-FLIPL appeared to be largely due to the interfering with recruitment of caspase 8 into the DISC and reducing local concentrations of procaspase 8 for its autoproteolytic cleavage and activation.17, 19, 39, 40 To determine how reduced CaM binding affects c-FLIPL and caspase 8 recruitment to the DISC, we performed IP of the DISC after Fas activation. Less c-FLIPL H204 mutant was in the DISC compared with WT c-FLIPL (Figure 4), which supports our previous hypothesis that disrupted CaM-c-FLIPL binding may cause a conformational change in c-FLIPL and decrease its recruitment into the DISC.25 The amount of caspase 8 recruited into the DISC decreased in c-FLIPL-overexpressing cells (Figure 4), suggesting that inhibition of caspase 8 activation by c-FLIPL was mainly through reducing caspase 8 in the DISC. Activated caspase 8 (p43/41) in the DISC appeared at 30 min in control and c-FLIPL H204 mutant cells, which was earlier than the 60 min seen in WT c-FLIPL cells (Figure 4). Our previous publication has demonstrated the highest CaM-c-FLIPL binding in response to CH-11 stimulation occurred at 30 min and returned to the basal level at 60 min in cholangiocarcinoma cells.25 That could explain why the largest difference between the effects of WT c-FLIPL and H204 mutant on caspase 8 activation was observed at 30 min. A proposed mechanism whereby CaM/FLIP binding modulates Fas-mediated apoptosis is shown in Figure 6. WT c-FLIPL competes with procaspase 8 binding to FADD and thus decreases caspase 8 recruitment to the DISC and its activation, thus inhibiting Fas-mediated apoptosis. The H204 mutant in c-FLIPL reduces its binding to CaM, which results in less c-FLIPL recruited to the DISC. Consequently, more procaspase 8 is recruited to the DISC, forming high local concentrations, which leads to its autoproteolytic cleavage and activation, which in turn activates downstream signals and increases apoptosis.32, 39, 40

Proposed model of the effects of WT c-FLIPL and H204 mutant on Fas-mediated DISC formation. The model depicts the Fas-mediated DISC formation and apoptotic signaling affected by WT c-FLIPL and H204 mutant in cholangiocarcinoma cells. WT c-FLIPL competes with procaspase 8 binding to FADD, thus decreasing caspase 8 recruitment to the DISC and its activation, which inhibits Fas-mediated apoptosis. The H204 mutant in c-FLIPL has reduced binding to CaM, which results in less c-FLIPL recruited to the DISC. Consequently, more procaspase 8 is recruited to the DISC, forming high local concentrations, which leads to its autoproteolytic cleavage and activation, which in turn activates downstream signals and increases apoptosis.
Fas is well characterized as a death receptor in the apoptotic machinery, but activation of Fas has also been shown to induce cell proliferation and tissue regeneration.41, 42 Recently, c-FLIP has been implicated in signaling alternative pathways, linking the Fas receptor to the NF-κB, JNK, MAPK and ERK pathways.25, 43, 44, 45, 46 These studies link the Fas-signaling pathway to survival pathways in addition to its well-known role in apoptosis. Our studies provide new evidence supporting the concept that the Fas-CaM-FLIP arm of the DISC might be important in modulating both apoptotic and survival signaling in response to Fas activation. Further characterizing the nature of CaM-FLIP interaction with respect to other death receptors such as DR4, DR5 and TNF-R1, may provide new therapeutic targets for cancer therapy.
References
Cohen GM . Caspases: the executioners of apoptosis. Biochem J 1997;326:1–16.
Pawar P, Ma L, Byon CH, et al. Molecular mechanisms of tamoxifen therapy for cholangiocarcinoma: role of calmodulin. Clin Cancer Res 2009;15:1288–1296.
Pan G, Vickers SM, Pickens A, et al. Apoptosis and tumorigenesis in human cholangiocarcinoma cells. Involvement of Fas/APO-1 (CD95) and calmodulin. Am J Pathol 1999;155:193–203.
Chen Y, Xu J, Jhala N, et al. Fas-mediated apoptosis in cholangiocarcinoma cells is enhanced by 3,3′-diindolylmethane through inhibition of AKT signaling and FLICE-like inhibitory protein. Am J Pathol 2006;169:1833–1842.
Ahn EY, Pan G, Oh JH, et al. The combination of calmodulin antagonists and interferon-γ induces apoptosis through caspase-dependent and -independent pathways in cholangiocarcinoma cells. Am J Pathol 2003;163:2053–2063.
Vickers SM, Jhala NC, Ahn EY, et al. Tamoxifen (TMX)/Fas induced growth inhibition of human cholangiocarcinoma (HCC) by g interferon (IFN-g). Ann Surg 2002;235:872–878.
Ahn EY, Pan G, Vickers SM, et al. IFN-gamma upregulates apoptosis-related molecules and enhances Fas-mediated apoptosis in human cholangiocarcinoma. Int J Cancer 2002;100:445–451.
Que FG, Phan VA, Phan VH, et al. Cholangiocarcinomas express Fas ligand and disable the Fas receptor. Hepatology 1999;30:1398–1404.
Jing G, Yuan K, Turk AN, et al. Tamoxifen enhances therapeutic effects of gemcitabine on cholangiocarcinoma tumorigenesis. Lab Invest 2011;91:896–904.
Chaigne-Delalande B, Moreau JF, Legembre P . Rewinding the DISC. Arch Immunol Ther Exp (Warsz) 2008;56:9–14.
Baumler C, Duan F, Onel K, et al. Differential recruitment of caspase 8 to cFlip confers sensitivity or resistance to Fas-mediated apoptosis in a subset of familial lymphoma patients. Leuk Res 2003;27:841–851.
Irmler M, Thome M, Hahne M, et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997;388:190–195.
Thome M, Schneider P, Hofmann K, et al. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 1997;386:517–521.
Shu HB, Halpin DR, Goeddel DV . Casper is a FADD- and caspase-related inducer of apoptosis. Immunity 1997;6:751–763.
Peter ME, Krammer PH . The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ 2003;10:26–35.
Chang DW, Xing Z, Pan Y, et al. c-FLIP(L) is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J 2002;21:3704–3714.
Scaffidi C, Schmitz I, Krammer PH, et al. The role of c-FLIP in modulation of CD95-induced apoptosis. J Biol Chem 1999;274:1541–1548.
Dohrman A, Russell JQ, Cuenin S, et al. Cellular FLIP long form augments caspase activity and death of T cells through heterodimerization with and activation of caspase-8. J Immunol 2005;175:311–318.
Thome M, Tschopp J . Regulation of lymphocyte proliferation and death by FLIP. Nat Rev Immunol 2001;1:50–58.
Babu YS, Sack JS, Greenhough TJ, et al. Three-dimensional structure of calmodulin. Nature 1985;315:37–40.
Dowd DR, MacDonald PN, Komm BS, et al. Evidence for early induction of calmodulin gene expression in lymphocytes undergoing glucocorticoid-mediated apoptosis. J Biol Chem 1991;266:18423–18426.
Gerner C, Frohwein U, Gotzmann J, et al. The Fas-induced apoptosis analyzed by high throughput proteome analysis. J Biol Chem 2000;275:39018–39026.
Yuan K, Jing G, Chen J, et al. Calmodulin mediates FAS-induced FADD-independent survival signaling in pancreatic cancer cells via activation of Src-ERK. J Biol Chem 2011;286:24776–24784.
Ahn EY, Lim ST, Cook WJ, et al. Calmodulin binding to the Fas death domain. Regulation by Fas activation. J Biol Chem 2004;279:5661–5666.
Pawar PS, Micoli KJ, Ding H, et al. Calmodulin binding to cellular FLICE-like inhibitory protein modulates Fas-induced signaling. Biochem J 2008;412:459–468.
Knuth A, Gabbert H, Dippold W, et al. Biliary adenocarcinoma. Characterisation of three new human tumor cell lines. J Hepatol 1985;1:579–596.
Ustundag Y, Bronk SF, Gores GJ . Proteasome inhibition-induces endoplasmic reticulum dysfunction and cell death of human cholangiocarcinoma cells. World J Gastroenterol 2007;13:851–857.
Sacks DB, Porter SE, Ladenson JH, et al. Monoclonal antibody to calmodulin: development, characterization, and comparison with polyclonal anti-calmodulin antibodies. Anal Biochem 1991;194:369–377.
Byon CH, Javed A, Dai Q, et al. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signalling. J Biol Chem 2008;283:15319–15327.
Chen J, Sun Y, Mao X, et al. RANKL up-regulates brain-type creatine kinase via poly(ADP-ribose) polymerase-1 during osteoclastogenesis. J Biol Chem 2010;285:36315–36321.
Wu X, Wakefield JK, Liu H, et al. Development of a novel trans-lentiviral vector that affords predictable safety. Mol Ther 2000;2:47–55.
Chen Y, Pawar P, Pan G, et al. Calmodulin binding to the Fas-mediated death-inducing signaling complex in cholangiocarcinoma cells. J Cell Biochem 2008;103:788–799.
Beneteau M, Daburon S, Moreau JF, et al. Dominant-negative Fas mutation is reversed by down-expression of c-FLIP. Cancer Res 2007;67:108–115.
Legembre P, Barnhart BC, Peter ME . The relevance of NF-kappaB for CD95 signaling in tumor cells. Cell Cycle 2004;3:1235–1239.
Rhoads AR, Friedberg F . Sequence motifs for calmodulin recognition. FASEB J 1997;11:331–340.
O’Day DH . CaMBOT: profiling and characterizing calmodulin-binding proteins. Cell Signalling 2003;15:347–354.
Irmler M, Thome M, Hahne M, et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997;388:190–195.
Golks A, Brenner D, Fritsch C, et al. c-FLIPR, a new regulator of death receptor-induced apoptosis. J Biol Chem 2005;280:14507–14513.
Kischkel FC, Hellbardt S, Behrmann I, et al. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a deathinducing signaling complex (DISC) with the receptor. EMBO J 1995;14:5579–5588.
Salvesen GS, Dixit VM . Caspase activation: The induced-proximity model. Proc Natl Acad Sci USA 1999;96:10964–10967.
Legembre P, Barnhart BC, Zheng L, et al. Induction of apoptosis and activation of NF-kappaB by CD95 require different signaling thresholds. EMBO Rep 2004;5:1084–1089.
Desbarats J, Newell MK . Fas engagement accelerates liver regeneration after partial hepatectomy. Nat Med 2000;6:920–923.
Hu WH, Johnson H, Shu HB . Activation of NF-kappaB by FADD, Casper, and caspase-8. J Biol Chem 2000;275:10838–10844.
Chaudhary PM, Jasmin A, Eby MT, et al. Modulation of the NFkappa B pathway by virally encoded death effector domains-containing proteins. Oncogene 1999;18:5738–5746.
Kataoka T, Budd RC, Holler N, et al. The caspase-8 inhibitor FLIP promotes activation of NF-kappaB and Erk signaling pathways. Curr Biol 2000;10:640–648.
Tran SE, Holmstrom TH, Ahonen M, et al. MAPK/ERK overrides the apoptotic signaling from Fas, TNF, and TRAIL receptors. J Biol Chem 2001;276:16484–16490.
Acknowledgements
This work was supported by VA Merit Review Award (JMM).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
Calmodulin (CaM) binds to c-FLIPL in a Ca2+-dependent manner. Deletion of the CaM-binding region increases Fas-mediated apoptosis and decreases tumorigenesis of cholangiocarcinoma cells. The H204 residue is responsible for c-FLIPL binding to CaM, which mediates the anti-apoptotic function of c-FLIPL through recruitment of caspase 8 into the death-inducing signaling complex.
Rights and permissions
About this article
Cite this article
Jing, G., Yuan, K., Liang, Q. et al. Reduced CaM/FLIP binding by a single point mutation in c-FLIPL modulates Fas-mediated apoptosis and decreases tumorigenesis. Lab Invest 92, 82–90 (2012). https://doi.org/10.1038/labinvest.2011.131
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2011.131

{kind=link}
{kind=link}