
Abstract
Liver fibrosis due to hepatic stellate cell (HSC) activation represents a common response to chronic liver injury. PTK787/ZK222584 (PTK/ZK) is a pan-VEGFR tyrosine kinase inhibitor. The aim of this study was to examine the effect of PTK/ZK in liver fibrosis. In primary HSCs, PTK/ZK inhibited the expression of α-smooth muscle actin (α-SMA), collagen, tissue inhibitor of metalloproteinase-1 (TIMP-1), as well as cell proliferation, migration and actin filament formation. PTK/ZK-induced apoptosis of HSCs, which was correlated with increased caspase-3 activation and suppressed Bcl-2 expression. PTK/ZK also induced cell cycle arrest, accompanied by increasing the expression of p27Kip1 and downregulation of cyclin D1 and cyclin E. PTK/ZK significantly inhibited vascular endothelial growth factor (VEGF) expression, as well as VEGF-simulated cell proliferation and phosphorylation of Akt in activated HSCs. In a murine fibrotic liver, PTK/ZK attenuated collagen deposition and α-SMA expression in carbon tetrachloride-induced fibrosis in both a ‘prevention’ and ‘treatment’ dosing scheme. These beneficial effects were associated with reduced phosphorylation of Akt and suppressed mRNA expression of procollagen-(I), TIMP-1, matrix metalloproteinase-9 and CD31. These findings provide novel insights into the potential value of blocking VEGF signaling by a small molecule tyrosine kinase inhibitor in treating hepatic fibrosis.
Similar content being viewed by others

Main
Chronic liver injury from chronic hepatitis, alcohol, cholestasis, drugs and a range of insults typically leads to liver fibrosis; its end stage, cirrhosis, is a major clinical problem worldwide because of life-threatening complications of portal hypertension, liver failure and hepatocellular carcinoma. Therapeutic efforts aim to halt or diminish the progression of liver fibrosis and thus prevent the development of liver cirrhosis and hepatocellular carcinoma. Unfortunately, there is currently no effective drug for liver fibrosis.
Hepatic fibrosis is characterized by altered extracellular matrix (ECM), consisting of a net increased synthesis of collagens, especially type I, and decreased matrix degradation catalyzed by the activity of matrix metalloproteinases (MMPs).1 Decreased activity of ECM-removing MMPs is mediated in part by enhanced expression of tissue inhibitors of metalloproteinases (TIMPs).1
The activated hepatic stellate cell (HSC) is primarily responsible for the synthesis of the excess ECM during liver fibrosis.2 In response to liver damage, HSC transforms from a quiescent vitamin A-rich cell type to an activated myofibroblast (α-smooth muscle actin (α-SMA)-expressing) phenotype, acquiring fibrogenic properties, secretion of proinflammatory cytokines, proliferation, migration and contractile capacity.3, 4, 5, 6, 7 Moreover, recovery from established experimental fibrosis can occur through the apoptosis of HSCs, and is associated with reductions in liver collagen and decreased expression of TIMP-1.8
Recent evidence indicates that angiogenesis is of central importance for many pathological events including fibrogenesis.9 Of those angiogenic modulating factors, vascular endothelial growth factor (VEGF) is the most potent. VEGF-induced angiogenesis may also contribute to wound repair, including hepatic fibrogenesis.10 The effects of VEGF are mediated mainly through two high affinity tyrosine kinase receptors, VEGF receptor-1 (Flt-1) and VEGF receptor-2 (Flk-1/KDR).11 Recently, Yoshiji et al12 reported that VEGF signaling is involved in the development of liver fibrosis. Moreover, activated HSCs express VEGF and VEGF receptors in the liver after carbon tetrachloride (CCl4) and may be responsive to hypoxia by expressing VEGF in vitro.13 However, downstream pathways of VEGF signaling in HSCs have not been evaluated.
Because of its role in fibrosis, inhibiting VEGF signal transduction provides an opportunity for therapeutic intervention. Indeed, blockage of VEGF receptors by specific neutralizing antibodies significantly attenuates the progression of liver fibrosis.12 An emerging therapeutic strategy aims to generate small molecule inhibitors of receptor tyrosine kinase domains. PTK787/ZK222584 (PTK/ZK), a potent pan-VEGFR TKI, inhibits angiogenesis, tumor growth and metastasis formation in experimental carcinoma models.14, 15 PTK/ZK has displayed activity in early clinical trials.16, 17 In this study, we examined the effects of PTK/ZK on the development of liver fibrosis induced by CCl4. Direct effects of PTK/ZK on fibrosis as well as HSC responses on VEGF-mediated signaling were investigated.
MATERIALS AND METHODS
Drug
PTK/ZK (succinate salt) was provided by Novartis Pharma AG (Basel, Switzerland). For in vitro assays, a stock solution of 50 mM PTK/ZK was prepared in DMSO. PTK/ZK (dihydrochloride salt) was synthesized as previously described.18 For in vivo studies, the dihydrochloride salt was dissolved in distilled water.
HSC Isolation and Culture
HSCs were purified from normal rats, the nonparenchymal cell suspension by a single step density gradient centrifugation with Nycodenz, characterized and cultured as described in detail previously.19 Experimental manipulations were performed with cells at passages 4–7.
Animals and Administration of CCl4
Male Balb/c mice, aged 6 weeks, were purchased from Laboratory Animal Unit. The research protocol was approved by the Institutional Ethics Committee. The experimental plan included a ‘prevention’ regimen and ‘treatment’ regimen. All fibrotic experimental groups received CCl4 (2 ml/kg per body weight dissolved in 150 μl of olive oil) twice per week. In the ‘prevention’ regimen, the first group (sham control) received olive oil injection, the second group received CCl4 injection for 6 weeks whereas animals in the third group were injected with CCl4 concurrently with administration with PTK/ZK p.o. (50 mg/kg) daily for 6 weeks. In the ‘treatment’ regimen, the first group (sham control) received olive oil injections. The second group was administered CCl4 for 2 or 4 weeks. The third group was administered CCl4 for 2 or 4 weeks and then administered PTK/ZK p.o. (50 mg/kg) daily for 6 weeks, together with CCl4 injection. A fourth group was injected with CCl4 for 8 or 10 weeks. At the time of killing, hepatectomy was performed and liver tissue samples were either frozen immediately in liquid nitrogen and stored at −70°C or fixed in 10% buffered formalin and embedded in paraffin.
Histological and Immunohistochemical Studies
Liver samples were formalin-fixed, paraffin-embedded, sectioned at 4 μm and processed routinely for Sirius red staining.20 Collagen stained with Sirius red was quantitated in six randomly selected areas per sample at a magnification of × 40. The area of positive staining was quantified using a computer-aided image analysis system NIS-Elements Advanced Research (Nikon, Tokyo, Japan). Immunohistochemical staining of α-SMA was performed as previously described.21 Morphometric quantification of α-SMA-positive area in liver specimens was determined as described before.
Serum Chemistries
Serum albumin (ALB), total bilirubin (TBil), alanine aminotransferase (ALT), aspartate aminotransferase (AST) were measured using standard laboratory assays. Serum hyaluronic acid (HA) was assayed using the HA test kit (Corgenix UK Ltd, Peterborough, Cambridgeshire, UK) according to the manufacturer's instructions.
Effects of PTK/ZK on HSC Proliferation and VEGF-Induced HSC Proliferation
Proliferation of HSC cells was measured by bromodeoxyuridine (BrdU) incorporation using BrdU labeling and detection kit (Roche Diagnostics Corporation, Indianapolis, IN, USA). Cells were plated at a density of 2 × 103 cells per well into 96-well plates and cultured overnight. PTK/ZK was added in serial dilutions in the medium containing 1% FCS and cell proliferation was measured by BrdU incorporation at 72 h. BrdU labeling solution was added, followed by incubation for another 16 h before fixation, addition of nucleases, anti-BrdU-POD and peroxidase substrate. The absorbance at 405 nm (with a reference wavelength at 490 nm) was measured using an ELISA plate reader (Molecular Devices Corp., Sunnyvale, CA, USA). For the effect of VEGF-induced HSC proliferation, HSCs were cultured in 96-well plates coated with acid-soluble type I collagen from rat tail tendon overnight,22 followed by washing cells with PBS twice and replacing growth medium with medium (0.1% FCS) containing VEGF (0.1–50 ng/ml; R&D Systems, Minneapolis, MN, USA). Cell proliferation was measured by BrdU incorporation at 48 and 72 h. For the effect of PTK/ZK on VEGF-induced cell proliferation, PTK/ZK was added 3 h before VEGF (10 ng/ml). Cell proliferation was performed using BrdU incorporation method as described before.
HSC Migration Assay
The migratory capacity of HSCs was investigated by using a BioCoat Matrigel Invasion Chamber (Becton Dickinson, San Jose, CA, USA). Confluent HSCs at the top chamber were incubated in serum free medium for 24 h, and the lower chamber was filled with PTK/ZK at incremental concentrations. After incubation for 24 h, cells from the upper surface of membranes were completed removed with gentle swabbing. The remaining migrated cells on the lower surface of membranes were fixed and stained with hematoxylin and eosin. Cellular migration was determined by counting the number of stained cells on membranes in five randomly selected fields at high power. HSCs that migrated through the membrane were enumerated by flow cytometry as described before.23
Effect of PTK/ZK on HSC Actin Cytoskeleton
HSCs were treated with incremental concentrations of PTK/ZK for 24 h. HSCs were then fixed with formaldehyde (3.7%) for 5 min at room temperature, followed by permeabilization with 0.1% Triton X-100. Cells were stained with 50 μg/ml fluorescent phalloidin conjugate (Sigma, St Louis, MO, USA) for 40 min, and analyzed under a fluorescence microscope (Olympus BX-50).
Effect of PTK/ZK on α-SMA Expression on HSCs by Flow Cytometry
HSCs were removed from the culture dish and fixed in 1 ml 4% paraformaldehyde, pelleted and resuspended in 1 ml of saponin buffer (0.1%, w/v saponin plus 0.05% azide in PBS) for permeabilization. The cells were washed with flow cytometry buffer (ice-cold PBS containing 2% FBS and 0.05% sodium azide) and incubated with α-SMA antibody (Dako, Glostrup, Denmark) for 45 min at 4°C. After washing, the cells were incubated with anti-mouse PE (BD PharMingen, San Diego, CA, USA) for 30 min. Cells were then washed and percentage of cell expressing α-SMA were subjected to flow cytometry analysis by FACS calibur (Becton Dickinson) according to the method described before.15 Mouse IgG2a (Zymed Laboratories, South San Francisco, CA, USA) was included as a negative control.
Detection of Cell Apoptosis by Flow Cytometry
HSCs were treated with PTK/ZK at different concentrations for 24 in the medium containing 1% FCS. According to BD PharMingen's instructions, HSCs were labeled with Annexin V-FITC (BD PharMingen, San Jose, CA, USA) and propidium iodide (PI; Sigma) for flow cytometry analysis.
Cell Cycle Analysis by Flow Cytometry
HSCs were treated with PTK/ZK in the medium containing 1% FCS for 24 h. Cells were washed, fixed with ice-cold 70% ethanol and incubated in 800 μl PBS, 100 μl RNase (1 mg/ml; Sigma) and 20 μl PI 2 mg/ml for 30 min at 37°C, followed by flow cytometry analysis using FACS calibur (Becton Dickinson). The percentage of cells in the G0–G1 and G2–M phases was assessed by ModFit LT software (Verity Software House, Topsham, ME, USA).
Effect of PTK/ZK on VEGFRs Expression on HSCs by Flow Cytometry Analysis
HSCs were pretreated with PTK/ZK at various concentrations overnight before removing the cells from plastics and labeling for VEGFRs (Flt-1, Flk-1/KDR) antibodies. HSCs were incubated with Flk-1/KDR or Flt-1 antibodies (Santa Cruz Biotechnology) for 45 min at 4°C, washed with ice-cold PBS, and then incubated with anti-rabbit PE (BD PharMingen) for 30 min. Cells were washed and then subjected to flow cytometry analysis by FACS calibur (Becton Dickinson). Rabbit IgG (Zymed Laboratories) was included as a negative control.
Analysis of mRNA Expression
The mRNA expression of α1-(I)-procollagen, CD31, TIMP-1, MMP-9 and VEGF was evaluated by real-time PCR. Total RNA was isolated from liver tissues and HSCs using RNeasy Mini kit (Qiagen Inc., Hilden, Germany), according to the manufacturer's instruction. For cDNA synthesis, Taqman reverse transcription reagents were used as described in the manufacture's protocol (PE Applied Biosystems, Foster City, CA, USA). Quantitative real-time PCR was performed using Taqman Universal PCR Master Mix kit (PE Applied Biosystems) on a PE Applied Biosystems 7700 sequence detector. The amplification conditions on the PE Applied Biosystems 7700 sequence detector were the following: 2 min at 50°C, 10 min at 95°C, 40 cycles of 95°C for 15 s and 60°C for 1 min for all reactions. Relative gene expression was calculated using 18S as internal control.
Western Blot
For analysis of the expression of collagen-I, caspase-3, Bcl-2, p27Kip1 (p27), cyclin D1, cyclin E, Flt-1, Flk-1, HSCs were treated with PTK/ZK at incremental concentrations in the medium with 1% FCS for 24 h, washed twice with PBS before lysis. For examine the effect of PTK/ZK on VEGF induction of expression of Akt, HSCs were serum deprived overnight, and then were treated with PTK/ZK for 3 h, followed by stimulation with VEGF 10 ng/ml for 10 min before lysis. For liver samples from mice, tissues were homogenized and lysed using lysis buffer. Following blocking, membranes were immunoblotted with the following antibodies: type I collagen (Rockland Inc., Gillbertsville, PA, USA), p27 (Transduction Laboratory, Lexington, KY, USA), caspase-3, Bcl-2, cyclin D1, cyclin E, Akt and phospho-Akt (Cell Signaling), Flt-1 and Flk-1 (Santa Cruz Biotechnology) for 18 h at 4°C, followed by detection using HRP-conjugated secondary antibody (Santa Cruz Biotechnology; 1 h, room temperature). Immunoreactive protein bands were visualized by the ECL system (Amersham Biosciences).
Statistical Analysis
Continuous data were expressed as mean±s.e.m. One-way ANOVA or two-tailed Student's t-test was used where appropriate. A P-value <0.05 was considered statistically significant.
RESULTS
PTK/ZK Inhibits HSC Activation in Culture
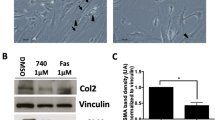
Isolated HSCs were incubated with PTK/ZK for 24 h at the concentration of 5 or 10 μM. HSC activation was evaluated by assessing intracellular expression of α-SMA using flow cytometry. The proportion of α-SMA-positive cells, determined by percentage of cells expressing α-SMA, was significantly decreased following PTK/ZK treatment (Figure 1a). The proliferation of HSCs is also a feature of cell activation and treatment with PTK/ZK for 3 days inhibited cell proliferation in a dose-dependent manner as assessed by BrdU incorporation (Figure 1b). Similarly, in HSCs treated with PTK/ZK, cells exhibited less migratory activity compared to untreated cells (Figure 1c, P<0.05). As shown in Figure 1d, activated HSCs exhibited well-organized bundles of F-actin stress fibers. PTK/ZK treatment led to a transformation of cell shape accompanied by the disruption of these fibers Figure 1d. As HSC activation is responsible for the increased synthesis and deposition of type I collagen in the liver, we determined whether PTK/ZK was involved in regulating α1 (I) collagen expression in activated HSCs. PTK/ZK treatment reduced type I collagen protein expression by western blot (Figure 1e). In addition, activated HSCs can also inhibit the activity of interstitial collagenases through upregulation of TIMP-1.24 Indeed, gene expression of TIMP-1 was elevated in activated HSCs and PTK/ZK treatment significantly reduced TIMP-1 mRNA (Figure 1f).

The effect of PTK/ZK on HSC activation in culture. (a) Expression of HSC activation marker α-SMA was significantly inhibited by PTK/ZK. Isolated HSCs were incubated with PTK/ZK for 24 h at the concentration of 5 or 10 μM, and labeled with α-SMA antibody for flow cytometry analysis. The percentage of α-SMA-positive cells was significantly decreased followed by PTK/ZK treatment. Data are mean±s.e. of three different experiments. (b) Effect of PTK/ZK on HSC proliferation. HSCs were cultured for 72 h in the presence of various concentrations of PTK/ZK. Proliferation was measured by BrdU incorporation assay. Results were representative of three experiments and each concentration was repeated six times in each experiment. (c) PTK/ZK inhibited migration of HSC measured by using Invasion Chamber as described in Materials and Methods section. HSCs were incubated in serum free medium for 24 h in the upper compartment of the chamber and were tested for migration in the lower compartment in the present of various concentrations of PTK/ZK. Cells migrated through to the lower compartment were quantified by cell counting. Values are expressed as a percentage of untreated control from three independent experiments. (d) PTK/ZK inhibited cytoskeleton rearrangement in activated HSCs. Fluorescence microscope shows changes in the cytoskeletal organization of rat HSCs after 24 h culture in the presence of PTK/ZK (10 μM). Fluorescent conjugates of phalloidin were used to label actin filaments. (e) HSCs were treated with incremental concentrations of PTK/ZK for 24 h. Analyzed by western blot, expression of collagen I was significantly inhibited by PTK/ZK. (f) Quantification of mRNA levels of TIMP-1. Total RNA was isolated from HSCs using RNeasy Mini kit, and TIMP-1 gene expression was measured by real-time PCR. PTK/ZK significantly suppressed TIMP-1 expression. TIMP-1 values were normalized to 18S mRNA values. Data are expressed as mean±s.e., *P<0.05.
PTK/ZK Induces Apoptosis of HSCs
PTK/ZK treatment of activated HSCs significantly increased the level of apoptosis as assessed by Annexin V-FITC and PI staining (Figure 2a). To investigate proapoptotic signaling pathways activated following PTK/ZK incubation, lysates of rat HSCs that had been treated for 24 h with PTK/ZK were analyzed by western blotting for caspase-3 and Bcl-2. As shown in Figure 2b, cleavage of caspase-3 protein was significantly increased after PTK/ZK treatment, whereas there was no significant change in the total caspase-3 protein. Reduced Bcl-2 protein expression was also observed in HSCs after PTK/ZK pretreatment.

The effect of PTK/ZK on HSC apoptosis. (a) HSCs were incubated with PTK/ZK at different concentrations for 24 h. Cells were harvested and double-stained for Annexin V-FITC and PI, apoptosis was quantified as Annexin V positive and PI negative. Representative of three experiments and each concentration was repeated twice in each experiment. Data are expressed as mean±s.e., *P<0.05. (b) The effect of PTK/ZK treatment on caspase-3 and Bcl-2 protein expression in HSCs. Western blot was performed on total lysates from HSCs treated with different concentrations of PTK/ZK for 24 h. The results of a representative study are shown. Two additional experiments yielded similar results.
PTK/ZK Induces Cell Cycle Arrest in Activated Rat HSCs
To further examine cell cycle effects, flow cytometric analysis of DNA content was performed on HSCs treated with PTK/ZK to investigate effects upon the cell cycle. PTK/ZK treatment significantly increased the number of cells in G0–G1 phase with a corresponding decrease in the number of cells in S and G2–M phases (Figure 3a). To determine the molecular basis of G1 arrest induced by PTK/ZK, we studied the expression of the cyclin-dependent kinase (CDK) inhibitor p27 in HSCs treated with incremental concentrations of PTK/ZK. As the G1 phase to S phase transition is primarily controlled by the D cyclins and cyclin E,25 we also examined the expression of cyclin D and cyclin E protein levels. PTK/ZK treatment at 5 or 10 μM significantly increased p27 expression and reduced cyclin D1 and cyclin E protein expression in HSCs pretreated with PTK/ZK at 5 or 10 μM (Figure 3b).

Effect of PTK/ZK on cell cycle distribution of HSCs. (a) HSCs were pretreated with PTK/ZK at various concentrations for 24 h, and then labeled with PI, followed by flow cytometry analysis. Compared to control untreated cells, PTK/ZK led to accumulation of cells in the G0–G1 phase and diminished distribution in the S and G2–M phases. (b) The effect of PTK/ZK treatment on p27, cyclin D1 and cyclin E protein expression in HSCs. Expression of the proteins in control HSCs or cells treated with PTK/ZK at different concentrations for 24 h was analyzed by western blot. The results of a representative study are shown. Two additional experiments yielded similar results.
PTK/ZK Reduces Expression of Flk-1, Flt-1 and VEGF in Cultured HSCs
As shown in Figure 4a and b and described previously,13 both Flk-1 and Flt-1 are expressed on activated HSCs. PTK/ZK treatment significantly inhibited Flk-1 and Flt-1 expression as measured by both the percentage of VEGF receptor positive cells and VEGF receptor expression levels (mean channel fluorescence) using flow cytometry (Figure 4a). However, only Flt-1 was inhibited by PTK/ZK as assessed by western blot, whereas there was no effect on Flk-1, contrast to results obtained by flow cytometry (Figure 4b). VEGF mRNA expression in activated HSCs was significantly suppressed by PTK/ZK (Figure 4c).

Effect of PTK/ZK on the expression of Flt-1, Flk-1 and VEGF on HSCs. (a) Activated HSCs were treated with PTK/ZK at various concentrations overnight. HSCs were then labeled with anti-Flt-1, or anti-Flk-1, followed by anti-rabbit PE, and analyzed by flow cytometry. Surface expression of Flt-1 and Flk-1 is presented as percentages of cells positive for the receptors, and also as mean channel fluorescence (MCF) on HSCs. Data are mean±s.e. from three experiments, *P<0.05. (b) HSCs were treated with PTK/ZK at indicated concentrations and cultured in the medium containing 1% FBS for 24 h. Cell lysates were subjected to western blot for analysis of Flt-1 and Flk-1 proteins. A representative study from three experiments is shown. (c) Effect of PTK/ZK on VEGF mRNA. HSCs were treated with PTK/ZK at the indicated concentrations before measurement of VEGF mRNA expression by real-time PCR. PTK/ZK incubation results in significant reduced VEGF mRNA expression. Data are presented as mean±s.e., n=3, *P<0.05.
PTK/ZK Suppresses VEGF Activity in Cultured HSCs
Consistent with a previous report,26 VEGF-induced proliferation of HSCs plated on type I collagen in a dose-dependent manner (Figure 5a), whereas PTK/ZK treatment significantly inhibited HSC proliferation induced by this cytokine (Figure 5b). As the PI3-kinase-Akt pathway reportedly involved in VEGF signaling and biological actions,27 we investigated whether VEGF activated the Akt pathway in HSCs. As shown in Figure 5c, VEGF promoted the phosphorylation of Akt. HSCs pretreated with PTK/ZK significantly blocked phosphorylation of Akt induced by VEGF (Figure 5c).

Effects of PTK/ZK on VEGF-induced HSC proliferation and Akt expression. (a) HSCs were cultured on plastic dishes precoated with type I collagen. VEGF was added at the indicated concentration and cells were incubated for 48 or 72 h. HSC proliferation was measured by BrdU incorporation as described in Materials and methods section. Data are mean±s.e. from three experiments and each concentration was repeated six times in each experiment, *P<0.05. (b) PTK/ZK at various concentrations was added to HSCs with or without VEGF (10 ng/ml). The proliferation of HSCs induced by VEGF was significantly inhibited by PTK/ZK in a dose-dependent manner. Data are presented as mean±s.e., n=3, *P<0.05. (c) PTK/ZK inhibited VEGF stimulated Akt pathway. HSCs were treated for 3 h with PTK/ZK where indicated, and were then stimulated with 10 ng/ml VEGF for 10 min. Cell lysates were analyzed for total Akt and phospho-Akt by western blot. The results of a representative study from three experiments are shown.
PTK/ZK Attenuates Liver Fibrosis In Vivo
After either 2 or 4 weeks of CCl4 injection, Sirius red staining revealed a marked increase in the extent of liver fibrosis, which was even more severe following 6, 8 or 10 weeks of CCl4 administration (Figure 6). PTK/ZK treatment significantly reduced the degree of liver fibrosis (Figure 6), an inhibitory effect that was more pronounced in the prevention regimen than in the treatment regimen, as expected. The antifibrotic properties of PTK/ZK in CCl4-treated mice were confirmed by assessing the effect of PTK/ZK on hepatic α-SMA protein expression in situ by immunohistochemistry. As shown in Figure 7, PTK/ZK dramatically suppressed α-SMA expression in CCl4-treated mice compared with the controls. To provide further evidence of the antifibrotic properties of PTK/ZK in CCl4-treated mice, we measured serum fibrosis marker HA. In both prevention and treatment regimens, HA was significantly lower compared to the control groups (Table 1). In addition, serum AST and ALT were significantly suppressed by treatment with PTK/ZK, whereas serum ALB and TBil levels did not change with the use of PTK/ZK (Table 1).

Effects of PTK/ZK on fibrosis in vivo. Photomicrographs of liver sections stained with Sirius red: (a) CCl4 injection for 6 weeks. (b) PTK/ZK administration started at the same time of CCl4 injection, and continued for 6 weeks. (c) CCl4 injection for 2 weeks. (d) CCl4 injection for 8 weeks. (e) After 2 weeks of CCl4, PTK/ZK was administered together with CCl4 for additional 6 weeks. (f) CCl4 injection for 4 weeks. (g) CCl4 injection for 10 weeks. (h) After 4 weeks of CCl4, PTK/ZK was administered together with CCl4 for additional 6 weeks. Mice were killed to assess the extent of liver fibrosis. Liver fibrosis was apparent by 2 weeks of CCl4, whereas PTK/ZK significantly suppressed liver fibrosis (original magnification × 200). (i) Quantitative analysis of liver fibrosis. Percent area of liver fibrosis was calculated using Sirius red staining as described in Materials and Methods section. Results are expressed as mean±s.e. of eight samples, *P<0.05 (original magnification × 40).

Hepatic immunohistochemical staining for α-SMA: for groups see caption of Figure 6. Many α-SMA-positive stellate cells are seen in livers of CCl4-induced mice, whereas significant decreased α-SMA-positive cells are present in livers of PTK/ZK-treated mice. α-SMA-positive areas in liver sections were quantified by NIS-Elements AR. Data are mean±s.e., n=8, *P<0.05 (original magnification × 200).
PTK/ZK Reduces α1-(I)-Procollagen, TIMP-1, MMP-9 and CD31 Gene Expression
We next investigated whether the reduced collagen deposition was associated with reduced mRNA expression. PTK/ZK administration markedly reduced α1-(I)-procollagen gene expression in the livers in both the prevention and treatment groups compared with the respective CCl4-treated control groups (Figure 8a). Given that MMPs and TIMPs are two major components of the ECM remodeling system, and that their activity closely correlates with the severity of liver fibrosis,28 we next compared TIMP-1 and MMP-9 gene expression in liver tissue samples obtained from CCl4-treated mice receiving PTK/ZK, and with those receiving CCl4 only. PTK/ZK treatment significantly downregulated TIMP-1 and MMP-9 gene expression (Figure 8b and c). Finally, we examined whether the inhibitory effects of PTK/ZK were also associated with suppression of neovascularization by evaluating CD31 gene expression in vivo, a marker of angiogenesis.29 PTK/ZK significantly suppressed CD31 gene expression compared with the control groups (P<0.01; Figure 8d). The inhibitory impact was more potent in the prevention groups that in the treatment groups.

mRNA expression of (a) procollagen, (b) TIMP-1, (c) MMP-9 and (d) CD31 in the livers of control and PTK/ZK-treated groups measured by real-time PCR. Total RNA was isolated from liver tissues using RNeasy Mini kit, and mRNA was quantitated by real-time PCR as described in Materials and Methods section. The expression was normalized as a ratio using 18S mRNA as a housekeeping gene. The expression of these molecules was significantly suppressed after PTK/ZK administration. The data represent mean±s.e. Each group consisted of eight mice. Statistically significant difference was compared with the groups of CCl4 injection only (*P<0.05).
PTK/ZK Inhibits Akt Activation In Vivo
The effects of PTK/ZK on Akt in vivo were evaluated by western blot. Figure 9 demonstrates that PTK/ZK markedly downregulated the expression of phosphorylated Akt in the livers of both prevention and treatment groups, whereas total Akt remained unchanged.

Effect of PTK/ZK treatment on Akt expression in the livers from mice received CCl4 with or without PTK/ZK administration. Phospho-Akt and total Akt protein expression were analyzed by western blot on total lysates from liver specimens of mice untreated (control) or treated with PTK/ZK at 50 mg/kg per day. Results showed were from four animals in each group. The relative expression of phospho-Akt was calculated by the ratio of the band intensity relative to β-actin.
DISCUSSION
PTK/ZK, a potent receptor tyrosine kinase inhibitor, was initially developed as an antiangiogenic agent that inhibits VEGFR. In our previous study, we demonstrated that PTK/ZK exerted a direct effect on tumor cells, in addition to endothelial cells.15 In this study, we examined whether activated HSC could also be the target of PTK/ZK.
The activated HSC is a major target for antifibrotic therapies based on its recognition as the major fibrogenic cell type of the injured liver.30, 31, 32 PTK/ZK inhibited HSC activation as indicated by inhibition of α-SMA expression and HSC proliferation and migration. Activated HSCs also display reorganized actin filaments and develop prominent cytoplasmic stress fibers,33 and these modifications were inhibited by PTK/ZK. PTK/ZK also significantly suppressed type I collagen expression, as well as inhibiting TIMP-1 expression.
We previously reported that PTK/ZK exerted a direct effect on tumor cells by inducing apoptosis and disrupting cell cycle progression.15 Similarly, PTK/ZK promoted apoptosis of HSCs coincident with increased activation of caspase-3, a central caspase in the apoptotic cascade.34 Moreover, our study also demonstrated for the first time that the expression levels of both Bcl-2 significantly decreased after PTK/ZK treatment. Recently, overexpression of Bcl-2 in activated HSCs resulted in resistance of apoptosis, which may contribute to the progression of fibrosis in chronic liver diseases.35 We also demonstrated that PTK/ZK-induced growth arrest of HSCs associated with G1 arrest and partial S and G2–M block. This effect was associated with an increase in p27 and a decrease in cyclin D1 and cyclin E expression. These changes in cell cycle proteins are likely to be important as the G1 to S transition is regulated by cyclin D and E. In cultured rat HSCs, expression of cyclin D1 and E correlates with cell proliferation.36
Furthermore, we assayed the expression of VEGF and its receptors in activated HSC, and its activity. Consistent with the previous reports,13, 37 VEGFR (Flt-1 and Flk-1) and VEGF are expressed in activated HSCs. PTK/ZK treatment significantly inhibited the surface expression of both Flt-1 and Flk-1 by flow cytometry and of only Flt-1 by western blot. Therefore, the effect of PTK/ZK on Flt-1 and Flk-1 might be divergent. PTK/ZK might indeed inhibit Flt-1 expression, whereas provoking Flk-1 internalization in HSCs. In addition, PTK/ZK not only reduced VEGF mRNA expression, but also inhibited VEGF stimulated proliferation of activated HSCs, and activation of Akt pathway, which is an important intracellular mediator of growth signals in HSCs. Akt activation stimulates HSC proliferation, mediates HSC survival and resistance to apoptosis, and increases collagen production by HSCs.38, 39
The antiproliferative effect of PTK/ZK on HSCs appears to be mediated through multiple mechanisms, including (1) inhibition of the Akt pathway, (2) induction of caspase-3 activity and inhibition of Bcl-2 expression, (3) an increase in the expression of CDK inhibitors p27 and decrease in the expression of cyclin D, cyclin E1. Our findings uncover a molecular link between PTK/ZK as a VEGF receptor tyrosine kinase inhibitor and Akt-mediated HSC proliferation, migration, collagen expression, apoptosis and cell cycle distribution. This also provided further evidence that the effect of PTK/ZK on HSC may also through targeting VEGFR and inhibition of its downstream target Akt, subsequently.
In this study, we have demonstrated that PTK/ZK also significantly blocks hepatic fibrosis in vivo induced by CCl4, based on its inhibitory effect on collagen deposition, α-SMA expression and serum markers. PTK/ZK not only reduced procollagen gene expression, but also suppressed TIMP-1 and MMP-9 and CD31 gene expression. TIMP-1 is involved in the pathogenesis of liver fibrosis and its increase in hepatic fibrosis leads to enhanced survival of activated stellate cells and reduced degradation of interstitial, or ‘scar’ matrix.40 TIMP-1 forms specific complexes with the proenzyme form of MMP-9. Therefore, the overexpression of MMP-9 in livers of CCl4 animals might be sequestering the available TIMP into MMP/TIMP complexes. Furthermore, MMP-9 is also contributes HSC activation.41 Angiogenesis and fibrosis commonly occur together in many disease states where neovascularization is believed to initiate the pathological cascade.42 Indeed, we found that CD31, a sensitive marker in endothelial cells, was markedly inhibited by PTK/ZK not only at onset of fibrogenesis induction, but also at 2 or 4 weeks after CCl4 injection, which more closely models the typical clinical situation.
The effect of PTK/ZK on AST and ALT after CCl4 injection indicates an additional activity in reducing inflammatory activity, a highly beneficial property of a therapy for hepatic injury and fibrosis. Moreover, consistent with the culture data, Akt phosphorylation was significantly inhibited in livers of mice that received PTK/ZK in addition to CCl4.
In conclusion, our data demonstrate that treatment with a receptor tyrosine kinase inhibitor PTK/ZK markedly attenuated development of liver fibrosis by direct suppression of HSC activation. In addition, some hepatoprotective activity of PTK/ZK is apparent based on the reduction in transaminases (Table 1), which offers an additional therapeutic advantage of this compound, as it could both improve hepatocyte integrity whereas reducing stellate cell activation as two independent but complementary activities. Importantly, PTK/ZK is capable of inhibiting liver fibrosis and blocking VEGF-regulated HSC activation. Our study provides the rationale for the therapeutic trials of PTK/ZK alone or in combination with other antifibrotic therapies for the treatment of liver fibrosis.
References
Bataller R, Brenner DA . Liver fibrosis. J Clin Invest 2005;115:209–218.
Brenner DA, Waterboer T, Choi SK, et al. New aspects of hepatic fibrosis. J Hepatol 2000;32:32–38.
Milani S, Herbst H, Schuppan D, et al. Procollagen expression by nonparenchymal rat liver cells in experimental biliary fibrosis. Gastroenterology 1990;98:175–184.
Marra F . Hepatic stellate cells and the regulation of liver inflammation. J Hepatol 1999;31:1120–1130.
Maher JJ, Bissell DM, Friedman SL, et al. Collagen measured in primary cultures of normal rat hepatocytes derives from lipocytes within the monolayer. J Clin Invest 1988;82:450–459.
Friedman SL, Rockey DC, McGuire RF, et al. Isolated hepatic lipocytes and Kupffer cells from normal human liver: morphological and functional characteristics in primary culture. Hepatology 1992;15:234–243.
Friedman SL . Mechanisms of hepatic fibrogenesis. Gastroenterology 2008;134:1655–1669.
Iredale JP, Benyon RC, Pickering J, et al. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest 1998;102:538–549.
Yoshiji H, Kuriyama S, Fukui H . Angiotensin-I-converting enzyme inhibitors may be an alternative anti-angiogenic strategy in the treatment of liver fibrosis and hepatocellular carcinoma. Possible role of vascular endothelial growth factor. Tumour Biol 2002;23:348–356.
Nissen NN, Polverini PJ, Koch AE, et al. Vascular endothelial growth factor mediates angiogenic activity during the proliferative phase of wound healing. Am J Pathol 1998;152:1445–1452.
Kanno S, Oda N, Abe M, et al. Roles of two VEGF receptors, Flt-1 and KDR, in the signal transduction of VEGF effects in human vascular endothelial cells. Oncogene 2000;19:2138–2146.
Yoshiji H, Kuriyama S, Yoshii J, et al. Vascular endothelial growth factor and receptor interaction is a prerequisite for murine hepatic fibrogenesis. Gut 2003;52:1347–1354.
Ankoma-Sey V, Wang Y, Dai Z . Hypoxic stimulation of vascular endothelial growth factor expression in activated rat hepatic stellate cells. Hepatology 2000;31:141–148.
Drevs J, Hofmann I, Hugenschmidt H . Effects of PTK787/ZK 222584, a specific inhibitor of vascular endothelial growth factor receptor tyrosine kinases, on primary tumor, metastasis, vessel density, and blood flow in a murine renal cell carcinoma model. Cancer Res 2000;60:4819–4824.
Liu Y, Poon RT, Li Q, et al. Both antiangiogenesis- and angiogenesis-independent effects are responsible for hepatocellular carcinoma growth arrest by tyrosine kinase inhibitor PTK787/ZK222584. Cancer Res 2005;65:3691–3699.
Morgan B, Thomas AL, Drevs J, et al. Dynamic contrast-enhanced magnetic resonance imaging as a biomarker for the pharmacological response of PTK787/ZK 222584, an inhibitor of the vascular endothelial growth factor receptor tyrosine kinases, in patients with advanced colorectal cancer and liver metastases: results from two phase I studies. J Clin Oncol 2003;21:3955–3964.
Klem J . Current studies with PTK787, an oral inhibitor of vascular endothelial growth factor in colorectal cancer. Clin Colorectal Cancer 2003;3:147–149.
Bold G, Altmann KH, Frei J, et al. New anilinophthalazines as potent and orally well absorbed inhibitors of the VEGF receptor tyrosine kinases useful as antagonists of tumor-driven angiogenesis. J Med Chem 2000;43:2310–2323.
Schafer S, Zerbe O, Gressner AM . The synthesis of proteoglycans in fat-storing cells of rat liver. Hepatology 1987;7:680–687.
Sakaida I, Uchida K, Matsumura Y, et al. Interferon gamma treatment prevents procollagen gene expression without affecting transforming growth factor-beta1 expression in pig serum-induced rat liver fibrosis in vivo. J Hepatol 1998;28:471–479.
Sakaida I, Matsumura Y, Akiyama S, et al. Herbal medicine Sho-saiko-to (TJ-9) prevents liver fibrosis and enzyme-altered lesions in rat liver cirrhosis induced by a choline-deficient L-amino acid-defined diet. J Hepatol 1998;28:298–306.
Bissell DM, Guzelian PS . Phenotypic stability of adult rat hepatocytes in primary monolayer culture. Ann NY Acad Sci 1980;349:85–98.
Wang XM, Yu DM, McCaughan GW, et al. Fibroblast activation protein increases apoptosis, cell adhesion, and migration by the LX-2 human stellate cell line. Hepatology 2005;42:935–945.
Arthur MJ . Collagenases and liver fibrosis. J Hepatol 1995;22:43–48.
Reed SI . Control of the G1/S transition. Cancer Surv 1997;29:7–23.
Ankoma-Sey V, Matli M, Chang KB, et al. Coordinated induction of VEGF receptors in mesenchymal cell types during rat hepatic wound healing. Oncogene 1998;17:115–121.
Wu LW, Mayo LD, Dunbar JD, et al. Utilization of distinct signaling pathways by receptors for vascular endothelial cell growth factor and other mitogens in the induction of endothelial cell proliferation. J Biol Chem 2000;275:5096–5103.
Benyon RC, Arthur MJ . Extracellular matrix degradation and the role of hepatic stellate cells. Semin Liver Dis 2001;21:373–384.
Geerts AM, De Vriese AS, Vanheule E, et al. Increased angiogenesis and permeability in the mesenteric microvasculature of rats with cirrhosis and portal hypertension: an in vivo study. Liver Int 2006;26:889–898.
Geerts A . History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin Liver Dis 2001;21:311–335.
Friedman SL, Roll FJ, Boyles J, et al. Hepatic lipocytes: the principal collagen-producing cells of normal rat liver. Proc Natl Acad Sci USA 1985;82:8681–8685.
Milani S, Herbst H, Schuppan D, et al. Procollagen expression by nonparenchymal rat liver cells in experimental biliary fibrosis. Gastroenterology 1990;98:175–184.
Rockey DC, Friedman SL . Cytoskeleton of liver perisinusoidal cells (lipocytes) in normal and pathological conditions. Cell Motil Cytoskeleton 1992;22:227–234.
Grutter MG . Caspases: key players in programmed cell death. Curr Opin Struct Biol 2000;10:649–655.
Novo E, Marra F, Zamara E, et al. Overexpression of Bcl-2 by activated human hepatic stellate cells: resistance to apoptosis as a mechanism of progressive hepatic fibrogenesis in humans. Gut 2006;55:1174–1182.
Kawada N, Ikeda K, Seki S, et al. Expression of cyclins D1, D2 and E correlates with proliferation of rat stellate cells in culture. J Hepatol 1999;30:1057–1064.
Corpechot C, Barbu V, Wendum D, et al. Hypoxia-induced VEGF and collagen I expressions are associated with angiogenesis and fibrogenesis in experimental cirrhosis. Hepatology 2002;35:1010–1021.
Reif S, Lang A, Lindquist JN, et al. The role of focal adhesion kinase-phosphatidylinositol 3-kinase-akt signaling in hepatic stellate cell proliferation and type I collagen expression. J Biol Chem 2003;278:8083–8090.
Lafdil F, Chobert MN, Couchie D, et al. Induction of Gas6 protein in CCl4-induced rat liver injury and anti-apoptotic effect on hepatic stellate cells. Hepatology 2006;44:228–239.
Murphy FR, Issa R, Zhou X, et al. Inhibition of apoptosis of activated hepatic stellate cells by tissue inhibitor of metalloproteinase-1 is mediated via effects on matrix metalloproteinase inhibition: implications for reversibility of liver fibrosis. J Biol Chem 2002;277:11069–11076.
Han YP, Zhou L, Wang J, et al. Essential role of matrix metalloproteinases in interleukin-1-induced myofibroblastic activation of hepatic stellate cell in collagen. J Biol Chem 2004;279:4820–4828.
Kalluri R, Sukhatme VP . Fibrosis and angiogenesis. Curr Opin Nephrol Hypertens 2000;9:413–418.
Acknowledgements
We thank Dr Jeremy Hughes (Phagocyte Laboratory, MRC Center for Inflammation Research, University of Edinburgh, UK), Professor Mien-Chie Hung (Department of Molecular and Cellular Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA) and Dr Nai-Sum Wong (Department of Biochemistry, The University of Hong Kong) for their valuable advice and comments. Xueming Qian for technical assistant.
This work was funded by Small Project Funding Programme of the University of Hong Kong, Shenzhen Bureau of Science, Technology and Information (Shenzhen Key Laboratory Advancement Scheme). NIH Grant DK56621 and the Feld Fibrosis Fund, to SLF.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liu, Y., Lui, E., Friedman, S. et al. PTK787/ZK22258 attenuates stellate cell activation and hepatic fibrosis in vivo by inhibiting VEGF signaling. Lab Invest 89, 209–221 (2009). https://doi.org/10.1038/labinvest.2008.127
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2008.127
Keywords
This article is cited by
-
Therapeutic targeting of the PDGF and TGF-β-signaling pathways in hepatic stellate cells by PTK787/ZK22258
Laboratory Investigation (2009)
