
Abstract
Polypoidal choroidal vasculopathy (PCV), a subtype of age-related macular degeneration (AMD) more frequently seen in East Asians, has both common and distinct clinical manifestations with typical neovascular AMD (tAMD). We aim to examine the extent to which common genetic variants are shared between these two subtypes. We performed the meta-analysis of association in a total of 1062 PCV patients, 1157 tAMD patients and 5275 controls of East Asian descent from the Genetics of AMD in Asians Consortium at the 34 known AMD loci. A total of eight loci were significantly associated with PCV, including age-related maculopathy susceptibility 2 (ARMS2)-HtrA serine peptidase 1 (HTRA1), complement factor H (CFH), C2-CFB-SKIV2L, CETP, VEGFA, ADAMTS9-AS2 and TGFBR1 (P<5 × 10−4) from the single-nucleotide polymorphism-based test and COL4A3 from the gene-based tests (Pgene=2.02 × 10−4). PCV and tAMD are genetically highly correlated (rg=0.69, P=4.68 × 10−3), with AMD known loci accounting for up to 36% variation. Weaker association for PCV was observed at ARMS2-HTRA1 (Pdif=4.39 × 10−4) and KMT2E-SRPK2(Pdif=4.43 × 10−3), compared with tAMD. Variants at CFH, CETP and VEGFA exhibited different association signals in East Asians, in contrast to those in European individuals. Our data suggest a substantially shared genetic susceptibility for PCV and tAMD, while also highlight the unique associations for PCV, which is useful in understanding the pathogenesis of PCV.
Similar content being viewed by others
Introduction
Age-related macular degeneration (AMD) is one of the leading causes of irreversible vision loss among people over 50 years of age. The number of affected individuals suffering from AMD by 2040 will substantially increase by an estimated 113 million, due to the rapid growing elderly population.1 Polypoidal choroidal vasculopathy (PCV), now considered a subtype of AMD, is more common in individuals of Asian descent, accounting for up to 60% of neovascular ‘wet’ AMD cases in East Asia.2, 3 Both PCV and typical neovascular AMD (tAMD) are type of choroidal neovascularization characterized by blood leakage and sub-retinal hemorrhage at the macular region.4 However, PCV patients have unique clinical features, such as a younger age of disease onset, more distinct polypoidal lesions, fewer sub-retinal deposits of drusen, a higher risk of massive subretinal hemorrhage, and poorer responses to standard anti-vascular endothelial growth factor (VEGF) treatment, as compared with tAMD.5
AMD is a complex disease with the heritability estimated at 45–70%.6 Genome-wide association studies have demonstrated that genetic variants within complement factor H (CFH),7, 8, 9 age-related maculopathy susceptibility 2 (ARMS2) and HtrA serine peptidase 1 (HTRA1)10, 11, 12, 13 are consistently associated with AMD risk in various ethnic groups. Recently, the International AMD Genomics Consortium identified a total of 34 AMD loci using a large collection of approximately 16 000 cases and 17 000 controls of European ancestry.14 The genome-wide genetic markers accounted for approximately 46.7% of variability in AMD risk. The genetic findings in AMD have led to the greatest advancement in the practice of medicine, that is, the introduction of antiangiogenic therapies for wet tAMD.15
In contrast to tAMD, previous genetic studies for PCV mainly employed smaller scale candidate gene approaches at selected single-nucleotide polymorphisms (SNPs). CFH and ARMS2-HTRA1 were confirmed to be the loci most strongly associated with PCV.16, 17, 18 Recent whole-genome exome sequencing identified a rare c.986A>G variant in the FGD6 gene significantly associated with PCV but not tAMD19. However, the extent to which common AMD genetic variations are shared or unique to PCV or tAMD in Asian populations is unknown. In this study, we analyzed SNPs data across the 34 AMD loci in 1062 PCV, 1157 tAMD cases and 5275 controls recruited across multiple sites of East Asia, from the Genetics of AMD in Asians Consortium. We sought to (i) identify independent association signals at the known locus with PCV and tAMD in East Asians, (ii) assess the similar or different association patterns at the associated loci and (iii) explore the genetic correlation and heritability using SNP data for these two clinical subtypes.
Materials and methods
Study populations and phenotyping
We included a total of 1062 PCV, 1157 tAMD cases and 5275 controls in Singapore, Hong Kong, Korea and Japan from the Genetics of AMD in Asians Consortium,20 all of them were East Asian ancestry. For the 34 AMD genetic loci discovered from participants of European descent,14 we examined their effects on the risk for PCV and tAMD. A detailed description of subject recruitment and phenotyping is provided in Supplementary Note and Supplementary Table S1. The diagnosis of neovascular AMD in each study was made based on detailed clinical examinations, including dilated fundus photography, fluorescent angiography, and optical coherence tomography of posterior segment. Indocyanine green angiography was performed to diagnose PCV.21 Cases with other macular diseases such as central serous chorioretinopathy, myopic choroidal neovascularization, or angioid streaks were excluded. All studies were approved by their respective local research ethics committees. Study protocols adhered to the tenets of the Declaration of Helsinki. The participants provided signed consent forms before the start of study.
Genotyping and imputation
We genotyped genomic DNA derived from whole blood samples of study participants using Illumina Human OmniExpress or Human Hap610-Quad beadchips20 (Supplementary Table S1). As Singapore cohort has SNPs genotyped on either OmniExpress or Hap610-Quad beadschip, only overlapped SNPs were used for further imputation as described in previous publication 20. For each sample collection, we conducted stringent quality control filters centrally. Duplicate DNA samples, individuals with low call rate (<95%), gender mismatch or ethnic outliers were excluded. SNPs were excluded if at low genotyping call rate (>5% missingness), monomorphic SNPs, with minor allele frequency<1%, or out of Hardy–Weinberg Equilibrium (P<10−6). After quality control filtering, the array genotypes of each study were pre-phasing with SHAPEIT version 2 (Delaneau et al.22) and were imputed using the East Asian population haplotypes (Han Chinese in Beijing, China and Japanese in Tokyo, Japan) from 1000 Genomes Project data23 as reference panels (build 37, phase 1 release, March 2012) with the software IMPUTE2 version 2.2 (Howie et al.24) (Supplementary Table S1). Imputed genotypes were called with an impute probability threshold of 0.9.. For this analysis, we included 3 827 370 SNPs that were present in all 4 sample collections.
Candidate AMD loci
We evaluated the association of SNPs from Genetics of AMD in Asians genotyped and imputed data across the 34 AMD loci identified by the International AMD Genomics Consortium.14 The 34 loci included previously 18 known loci and 16 newly identified loci involved in: complement pathways and immune response (CFH, C2-CFB-SKIV2L, C3, C9 and CF1), angiogenesis signaling pathways (VEGFA, TGFBR1 and ADAMTS9-AS2), lipid metabolism(CETP, APOE, LIPC and ABCA1), oxidative stress pathways or cell survival (ARMS2-HTRA1 and TNFRSF10A), extracellular matrix remodeling (COL8A1, COL4A3, MMP3 and CTRB1-CTRB2), immune function (PILRB-PILRA), retina dehydrogenase (RDH5) and other 14 loci (Supplementary Table S2).
Statistical analysis
For each individual cohort, we performed case-control association test under an additive model for each genotyped or imputed SNP at the 34 AMD loci with PCV or tAMD status as the binary outcome. The number of risk alleles is an ordinary variable (0, 1 and 2) for directly genotyped SNPs, or a continuous variable of allele dosage probability ranging from 0 to 2 for imputed SNPs. We constructed principal components using the software EIGENSTRAT version 2.0, (http://morgan.dartmouth.edu/Docs/eigenstrat-2.0/, Department of Genetics, Harvard Medical School, Boston, MA, USA) and included the top five principal components as covariates in the logistical regression as previously described.20 For each SNP, we applied PLINK version v1.07 (http://pngu.mgh.harvard.edu/purcell/plink/; Broad Institute of Harvard & MIT, Cambridge, MA, USA) for association test in individual study and combined the statistics of association test from each cohort using the fixed-effect meta-analysis with inverse-variance weighting using the software METAL version 2010-02-08.25
We extracted all association results of SNPs residing at the 34 AMD loci with the boundaries determined by the ranges of correlated variants (r2⩾0.5) with all independently associated AMD SNPs identified from the IMDGC consortium. Given that all 34 loci have a prior evidence of genome-wide significant association with AMD and a medium of ~100 SNPs tested at each locus, we considered P<5 × 10−4 as the threshold of significance. The difference in the strength of SNP associations between PCV and tAMD was tested in the logistic regression for the eleven associated regions.
At loci that exhibited evidence of association at P<5 × 10−4, we next performed sequential conditional analyses by adding the most strongly associated SNP into the regression model as a covariate and tested all remaining regional SNPs for association. We repeated this process until the strongest SNP showed a conditional P⩾5 × 10−4.
Pair-wise measures of linkage disequilibrium (LD) r2 were obtained from the 1000 Genome Project 2013 release and reported for European and Asian population separately in Table 1. The coordinates and variant identifiers are reported on the NCBI B37 (hg19) genome build. We presented the regional association plot using LocusZoom (http://locuszoom.sph.umich.edu/locuszoom/) and used the LD estimates from the 1000 Genomes East Asian samples (Han Chinese in Beijing, China+Japanese in Tokyo, Japan). Relationships between genotype and cis regulation of gene expression levels were assessed using expression quantitative trait locus associations obtained from GTExPortal database26.
Gene-based analyses
The gene-based test was performed using the meta-analysis SNP association results by VEGAS version 2 software (https://vegas2.qimrberghofer.edu.au/; QIMR Berghofer Medical Research Institute, Brisbane, Queensland, Australia), accounting for LD between SNPs using simulations from the multivariate normal distribution.27 We used the combined 1000 Genomes East Asian populations as the reference for LD estimates. To include all SNPs linked with a certain gene, we annotated SNPs within the gene and SNPs in LD (r2>0.8) attributing to each gene. For the 49 genes across the 34 loci, we used the threshold of P<1.0 × 10−3 (P=0.05/49) as the significance level for gene-based association tests.
Genetic-determined disease variance and genetic correlation estimates
We estimated the proportion of variances explained by the SNPs by calculating pseudo R2 for the logistic regression model using McFadden’s method24. We obtained the likelihood separately in each study for the full model (Lm) with a set of SNPs and covariates (top five principal components) used in the association analysis, and the reduced model in which only covariates were included (L0). McFadden’s R2 is defined as R2McF=1−ln (LM)/ln (L0), where ln (.) is the natural logarithm. We calculated the proportion of variances explained by SNPs (a subset of SNPs) in each study cohort with the average of the estimates reported.
To estimate the genetic correlation between PCV and tAMD, we applied the LDscore method28 using genome-wide association summary statistics for each phenotype. We followed the procedure for LD regression calculation at https://github.com/bulik/ldsc/wiki. After merging SNPs with the HapMap3 Asian samples, we had a total of 804 865 SNPs for the LD score regression analyses. LD score matrix was estimated from the 1000 Genomes Project Asian reference panel with a 1 cM sliding window, provided by Bulik-Sullivan.28 We estimated the genetic covariance by regressing the product of testing statistics (z-statistics) from each phenotype against the total sample sizes of study. The resulting regression slope was multiplied by the number of effective SNPs in the reference East Asian panel from the 1000 Genomes Project data.28 We calculated the genetic correlation rg using the software ldsc version 1.0.0 (URL: https://github.com/bulik/ldsc/wiki/; Broad Institute of MIT and Harvard, Cambridge, MA, USA). We also reported rg at the liability scale to account for disease prevalence, assuming the prevalence of PCV/tAMD at 0.3% in the general population1.
Power calculation
We used the software Quanto (v1.2.4; http://biostats.usc.edu/Quanto.html) for assessing the ad-hoc power to detect regional association for the known loci. The power for each SNP was calculated individually, using the corresponding allele frequency, independent risk effect estimated from current study. We assumed a 1:5 ratio between cases and controls, complete LD, a disease prevalence of 0.3%1 and a 5 × 10−4 type 1 error in an allelic test.
Results
Loci associated with both PCV and tAMD
Supplementary Table S1 summarizes the genotype and phenotyping information for the participants comprising 1062 PCV patients, 1152 tAMD patients and 5275 non-AMD controls. Among the 34 AMD loci identified in European populations from International AMD Genomics Consortium,14 7 loci exhibited evidence of association for PCV: ARMS2-HTRA1, CFH, C2-CFB-SKIV2L, CETP, VEGFA, DAMTS9-AS2 and TGFBR1 (P<5 × 10−4; Table 1; Supplementary Table S2). Of these, four loci (ARMS2-HTRA1, CFH, C2-CFB-SKIV2L and CETP) were also significantly associated with tAMD. Results of replication at all the 34 loci for PCV are presented in Supplementary Table S3. An additional four loci showed significant association with tAMD only at NRTN-FUT6-C3, KMT2E-SRPK2, SLC16A8 and COL8A1 (P<5 × 10−4; Table 1 and Supplementary Table S4). Their genetic effects on PCV, although not significant, were largely in the same direction with those of tAMD.
The greatest effects were observed at the ARMS2-HTRA1 locus (rs36212731; PCV, odds ratio (OR)=2.18, P=1.09 × 10−46; tAMD, OR=2.78, P=2.26 × 10−80). SNP rs36212731 was in strong LD with the European lead SNP rs62247658 (r2=0.97) and the commonly studied non-synonymous variant A69S (rs10490924, r2=0.98). No further independent associated variants were observed at this locus in Asians. Our data suggest that causal common variants at ARMS2-HTRA1 for PCV/tAMD are similar across different ethnic groups.
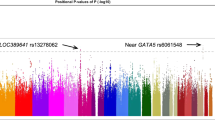
The most strongly associated SNP rs514591 in CFH (OR=1.87, P=1.45 × 10−26) was in strong LD (r2=0.96) with the largely investigated non-synonymous variant rs800292 (I62V; P=1.86 × 10−26). In East Asians, CFH rs514591 was moderately correlated with the lead SNP rs10922109 (r2=0.35) identified in European populations14 and weakly correlated with the widely studied non-synonymous rs1061170 (Y402H; r2=0.035; Figure 1a). Locus-wide conditional analysis pinpointed independent signal at rs3753394 in the promoter region (OR=1.41, P=1.83 × 10−10) and rs6689009 (OR=2.72, P=4.98 × 10−9; Supplementary Table S5). For tAMD, rs514591 also showed the strongest association (OR=1.75, P=5.70 × 10−24). The other two SNPs, rs3753394 and rs6689009, exhibited similar effects on tAMD compared to PCV. Both SNPs are intronic, whereas rs3753394 is associated with cis-acting expression of CFH in whole blood and is associated with cis-acting expression of CFHR3 in multiple tissues (Supplementary Table S6).

Regional association plots for PCV at the CFH, CETP and VEGFA loci. SNP number labeled in red represents the lead SNP identified from East Asians in current study, and those in black represents the lead SNP identified in Europeans from the International AMD Genomics Consortium (IAMDGC). A full color version of this figure is available at the Journal of Human Genetics journal online.
The strongest PCV-associated SNP, rs183130, at CETP (OR=1.40, P=3.07 × 10−7) was in high LD with the commonly studied SNP rs3764261 (r2=1.0) in Europeans. The low-frequency non-synonymous variant rs2303790 (D442G) also showed its independent association at the CETP locus (OR=1.84, P=2.71 × 10−6; Pcondi=2.50 × 10−4; Supplementary Table S5). Similar association patterns were noted for tAMD at rs183130 (P=4.31 × 10−5) and rs2303790 (P=3.98 × 10−4; Pcondi=8.60 × 10−3). SNP rs183130 is associated with cis-acting expression of CETP in artery (Supplementary Table S6). The strongest association signals identified in Europeans were at rs5817082, rs1864163 and rs17231506, whereas only rs17232506 had significant association with PCV and tAMD (Figure 1b).
The most significantly associated SNP rs401775 (P=3.89 × 10−8) at the C2-CFB-SKIV2L locus was in weak LD (r2=0.20) with the lead SNP rs429608 identified in Europeans (Supplementary Figure S1). Conditional analysis adjusting for rs429608 did not attenuate the association at rs401775 (Pcondi=7.15 × 10−6; Supplementary Table S5), suggesting that rs401775 could be an independent variant for PCV at the C2 locus.
Association differences for PCV and tAMD
We compared the strength of association signals for the two clinical subtypes at 14 lead SNPs across 11 associated loci. ARMS2-HTRA1 had an appreciable smaller effect size for PCV compared to tAMD (rs36212731; ORdif=0.81; Pdif=6.90 × 10−4; Table 1 and Figure 2). The KMT2E-SRPK2 locus showed a smaller association in PCV versus tAMD (rs35601145; ORdif=0.82, Pdif=4.43 × 10−3). SNP rs35601145 had the strongest association with tAMD (OR=1.22, P=2.08 × 10−4); it was in moderate LD (r2=0.68) with the lead SNP rs1142 in Europeans. In contrast to tAMD, none of SNPs at the KMT2E-SRPK2 locus was significantly associated with PCV (P⩾0.257; regional plots in Supplementary Figures S1 and S2).

Comparison of genetic effects for PCV and tAMD at associated loci. The circle represents the SNP at 11 loci as shown in Table 1. Red circles indicate the significant differences of association between PCV and tAMD at ARMS2-HTRA1 (Pdif=6.90 × 10−4) and KMT2E-SRPK2 (Pdif=4.43 × 10−3). A full color version of this figure is available at the Journal of Human Genetics journal online.
At the VEGFA locus, the PCV lead SNP rs3734693 (OR=1.32; P=1.29 × 10−6) was located within the transcript RP5-1120P11.1, 138 kb downstream from VEGFA rs943080, a marker previously showing strong association with AMD in Europeans but barely associated with PCV in our Asian samples (Figure 1c). These two SNPs were not in LD (r2=0.001). Conditional analysis indicated that rs3734693 was an independent signal from rs943080 (P=1.53 × 10−6). eQTL data suggest that rs3734693 is responsible for RNA gene expression of RP5-1120P11.1 (Supplementary Table S6). A weaker association was observed for tAMD at rs3734693 (OR=1.17, P=3.25 × 10−3; Table 1 and Supplementary Figure S1), whereas no statistically significant differences were found in the association between PCV and tAMD (ORdif=1.07; Pdif=0.285). Another susceptibility variant rs2295334 in C6orf223, previously identified to associate with neovascular AMD in East Asians,20 also exhibited similar association evidence for PCV and tAMD (PCV: P=2.38 × 10−4; tAMD: P=9.13 × 10−3; Pdif=0.280) in our samples.
Previously identified non-synonymous SNPs rs2230199 (R102G) and rs147859257 (K155Q) in C3 at the NRTN-FUT6-C3 locus from Europeans were rare in East Asian samples (minor allele frequency <0.005). We observed an association at rs12019136 within the NRTN-FUT6 region with tAMD (OR=1.44, P=1.45 × 10−4, Table 1); this SNP is about 880 kb downstream to C3 rs2230199 and these two SNPs were in low LD (r2=0.001; Supplementary Figure S1). No evidence of association was found at rs12019136 for PCV (OR=1.11; P=0.268) and a borderline statistical difference in the association between PCV and tAMD (ORdif=0.80; Pdif=0.061).
Gene-based analysis
We performed gene-based tests for 49 genes at 34 loci. There was a significant association at COL4A3 for PCV (PCV, Pgene=2.02 × 10−4; tAMD, Pgene=9.70 × 10−3; Table 2). Three loci had nominal significant association (Pgene<0.05) for PCV at TNFRSF10A, B3GALTL, and C20orf85, and two loci with tAMD: CNN2 and RORB. In addition, similar significance patterns for PCV and tAMD were observed for 11 loci (Pgene<0.05; Supplementary Table S7), except at COL8A1 (PCV, Pgene=0.592; tAMD, Pgene=0.333). Examining adjacent genes at the COL8A1 locus, we observed marginal significance at CMSS1 (Pgene=0.0173) and FILIP1L for tAMD (Pgene=0.0126), but not for PCV. Consistent with the SNP-based association test, we found only the NRTN and FUT6 genes at the FUT6-NRTN-C3 locus, but not C3, to be associated with tAMD.
SNP-determined disease variance and genetic correlation
We further evaluated the phenotypic variation explained by the top SNPs for PCV and tAMD in our East Asian participants. Using the four most strongly associated variants in ARMS2-HTRA1, CFH, CETP and C2-CFB-SKIV2L, we obtained the estimates of variances of 12.43% explained for PCV and 14.18% for tAMD (Figure 3). Adding another seven lead variants associated with PCV or tAMD (Table 1) and COl4A3 rs72975997 (Supplementary Table S3) increased the genetic heritability estimates to 22.20% for PCV and 23.57% for tAMD. The 34 genetic loci all together explained 36.84% disease variation for PCV and 37.30% for tAMD. At the individual variant level, lead SNP ARMS2-HTRA1 rs36212731 alone explained the largest variances (PCV: 5.25%; tAMD: 7.90%), followed by CFH rs514591 (PCV: 4.09%; tAMD: and 3.40%). Using all genome-wide SNPs, we calculated a genetic correlation between PCV and tAMD: rg=0.693 (s.e.=0.245, P=4.68 × 10−3) and at the liability scale: rg=0.699 (s.e.=0.245, P=4.60 × 10−3), indicating significantly shared genetic determinates of these two clinical subtypes.

Disease variance explained by known AMD loci. The phenotypic variation was estimated by using lead genetic variants at (1) 4 loci: ARMS2-HTRA1, CFH, C2, CETP, (2) 11 loci: variants at above 4 loci plus VEGFA, ADAMTS9-AS2, TGFBR1, NRTN-FUT6-C3, KMT2E-SRPK2, SLC16A8, COL8A1 and COL4A3, and 34 known AMD loci. A full color version of this figure is available at the Journal of Human Genetics journal online.
Discussion
Our study showed that common susceptibility SNPs at AMD known loci make an important contribution to PCV risk, demonstrating substantial sharing of genetic risk variants for PCV and tAMD. Eight AMD loci, including ARMS2-HTRA1, CFH, C2-CFB-SKIV2L, CETP, VEGFA, ADAMTS9-AS2, TGFBR1 and COL4A3 were associated with PCV, with the strongest associations at ARMS2-HTRA1 and CFH. The involvement of these loci highlighted the role of several common pathways for PCV: complement and immune response (CFH and C2-CFB-SKIV2), angiogenesis signaling pathways (VEGFA, TGFBR1 and ADAMTS9), lipid transport (CETP), oxidative stress pathways (ARMS2-HTRA1 and TNFRSF10A) and extracellular matrix (COL4A3). This study represents the most comprehensive genetic association study to compare PCV and tAMD in East Asian cohorts performed by the Genetics of AMD in Asians consortium.
We found 34 known AMD loci accounted for up to 36.8% of phenotypic variations for PCV, similar to those for tAMD (37.3%) in the same collection of Asian samples. Although 22 AMD loci were not significantly associated with PCV and tAMD after stringent multiple corrections, those SNPs together explained 14% phenotypic variation. Using genome-wide SNP data, our study first demonstrates that a high genetic correlation between these two clinical subgroups (rg=0.69), implying PCV and tAMD subtypes exhibiting a significant degree of genetic overlap. The empirical evidence of shared genetic etiology encouraged the investigation of common pathophysiologies for these related subphenotypes. Meanwhile, novel genetic variants, either common or rare, remain to be discovered for PCV in a much larger genome-wide association studies or whole-genome sequencing study.
Of the 11 AMD loci, majority exhibited similar effect sizes for PCV and tAMD, with few exceptions (Table 1 and Figure 1). These differences may correlate with the differences in the pathologic and clinical manifestations of PCV and tAMD. At the well-established ARMS2-HTRA1 locus, variants had a weaker association with PCV than with tAMD, which is in line with previous studies.29, 30 This could be due to the differences in the association of variants in ARMS2-HTRA1 between the two angiographic subtypes of PCV. The association at ARMS2 A69S was only seen for Type 1, not Type 2 PCV, in Japanese samples31, 32, suggesting genetic diversity for the PCV subtypes.
In addition to ARMS2-HTRA1, differential associations between PCV and tAMD were also noted at KMT2E-SRPK2, with a significant association only presenting in tAMD. SRPK2 encodes serine/arginine-rich protein-specific kinase 2. The inhibition of the protein SRPK1 and SRPK2 reduced angiogenesis in vivo; it has showed that knockdown of SRPK1 reduced VEGF-mediated angiogenesis in vivo in tumors33. The molecular mechanisms underlying the pathological and clinical manifestation of the two clinical subtypes remain to be elucidated.
We confirmed that a few loci (CFH, CETP and VEGFA) have different associated signals in East Asians for both PCV and tAMD, comparing with those reported in European ancestry individuals. This might be due to heterogeneity in allele frequencies, LD, or likely underlying different causal variants across ethnic groups. The strongest associations for PCV and tAMD at CFH locus were at rs514591, in complete LD with the functional variant rs800292 (I62V) in exome 2, and in moderate LD with rs3753394 in the promoter region. Both risk alleles of I62V and rs3753394 were twofold more frequent in Asians than in European populations. The established variant rs1061170 (Y402H), present at low frequencies (<5%) in Asians compared with Europeans (45%),29, 34 did not have an independent association signal in our samples. I62V and rs514591, located near a C3b-binding site, are involved in the binding affinity of C3b and thus likely to be the most important variants for AMD in the complement pathway in East Asians.
At the VEGFA locus, the strongest association was at rs3734693 within the transcript PR-1120P11, an independent variant from the lead VEGFA SNP rs943080 in Europeans. Previously, we also reported c6orf223 rs2295334, right adjacent to VEGFA, as the strongest signal for neovascular AMD in East Asians.20 The two SNPs, rs3734693 and rs2295334, were in moderate LD (r2=0.49) and likely attributed to the same underlying causal variants in East Asians.
Several limitations of our study warrant some attention. First, we have focused on common variants and rare variants (minor allele frequency <1%) are not investigated. Rare variants can be functional and effects are expected to vary across populations or environments. By assessing variants at the known loci, our study design only has 80% power to detect associations at allele frequency of 1% with at least 2.5-fold increased relative risk. A deep-sequencing study at the target regions will be needed to identify functional rare variants for PCV in individuals of Asian ancestry. In addition, the phenotypic variation estimates explained by genetic variants from our cases–control study, however, are likely to be inflated as the prevalence of PCV or tAMD is artificially higher in these selected cases and controls (cases to controls ratio: 1:5) than that in the general population (<1%).1 Furthermore, when the SNP minor allele frequency is nearer to 0, the estimated R2 tends to be larger using the McFadden method.35 Our estimates thus could be served as the upbound phenotypic variances explained by the SNPs examined. The figures, nevertheless, should be comparable between two subtypes as we used the same case-control study design and analytic approach.
In summary, we employed a large scale multi-centered case–control study to evaluate the genetic associations for PCV and tAMD in East Asians. Beyond demonstrating significantly shared genetic risk factors for PCV and tAMD, our results also highlight differences of association patterns for PCV. Our study adds to the understanding of the genetic architecture underlying PCV and tAMD susceptibility and may provide the opportunities for the medical intervention for PCV.
References
Wong, W. L., Su, X., Li, X., Cheung, C. M., Klein, R., Cheng, C. Y. et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob. Health 2, e106–e116 (2014).
Lim, T. H., Laude, A. & Tan, C. S. Polypoidal choroidal vasculopathy: an angiographic discussion. Eye (Lond) 24, 483–490 (2010).
Sho, K., Takahashi, K., Yamada, H., Wada, M., Nagai, Y., Otsuji, T. et al. Polypoidal choroidal vasculopathy: incidence, demographic features, and clinical characteristics. Arch. Ophthalmol. 121, 1392–1396 (2003).
Sharma, K., Sharma, N. K. & Anand, A. Why AMD is a disease of ageing and not of development: mechanisms and insights. Front. Aging Neurosci. 6, 151 (2014).
Laude, A., Cackett, P. D., Vithana, E. N., Yeo, I. Y., Wong, D., Koh, A. H. et al. Polypoidal choroidal vasculopathy and neovascular age-related macular degeneration: same or different disease? Prog. Retin. Eye Res. 29, 19–29 (2010).
Seddon, J. M., Cote, J., Page, W. F., Aggen, S. H. & Neale, M. C. The US twin study of age-related macular degeneration: relative roles of genetic and environmental influences. Arch. Ophthalmol. 123, 321–327 (2005).
Edwards, A. O., Ritter, R. 3rd, Abel, K. J., Manning, A., Panhuysen, C. & Farrer, L. A. Complement factor H polymorphism and age-related macular degeneration. Science 308, 421–424 (2005).
Klein, R. J., Zeiss, C., Chew, E. Y., Tsai, J. Y., Sackler, R. S., Haynes, C. et al. Complement factor H polymorphism in age-related macular degeneration. Science 308, 385–389 (2005).
Haines, J. L., Hauser, M. A., Schmidt, S., Scott, W. K., Olson, L. M., Gallins, P. et al. Complement factor H variant increases the risk of age-related macular degeneration. Science 308, 419–421 (2005).
Rivera, A., Fisher, S. A., Fritsche, L. G., Keilhauer, C. N., Lichtner, P., Meitinger, T. et al. Hypothetical LOC387715 is a second major susceptibility gene for age-related macular degeneration, contributing independently of complement factor H to disease risk. Hum. Mol. Genet. 14, 3227–3236 (2005).
Jakobsdottir, J., Conley, Y. P., Weeks, D. E., Mah, T. S., Ferrell, R. E. & Gorin, M. B. Susceptibility genes for age-related maculopathy on chromosome 10q26. Am. J. Hum. Genet. 77, 389–407 (2005).
Maller, J., George, S., Purcell, S., Fagerness, J., Altshuler, D., Daly, M. J. et al. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nat. Genet. 38, 1055–1059 (2006).
Dewan, A., Liu, M., Hartman, S., Zhang, S. S., Liu, D. T., Zhao, C. et al. HTRA1 promoter polymorphism in wet age-related macular degeneration. Science 314, 989–992 (2006).
Fritsche, L. G., Igl, W., Bailey, J. N., Grassmann, F., Sengupta, S., Bragg-Gresham, J. L. et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 48, 134–143 (2015).
Black, J. R. & Clark, S. J. Age-related macular degeneration: genome-wide association studies to translation. Genet. Med. 18, 283–289 (2016).
Ng, T. K., Chen, L. J., Liu, D. T., Tam, P. O., Chan, W. M., Liu, K. et al. Multiple gene polymorphisms in the complement factor h gene are associated with exudative age-related macular degeneration in chinese. Invest. Ophthalmol. Vis. Sci. 49, 3312–3317 (2008).
Lee, K. Y., Vithana, E. N., Mathur, R., Yong, V. H., Yeo, I. Y., Thalamuthu, A. et al. Association analysis of CFH, C2, BF, and HTRA1 gene polymorphisms in Chinese patients with polypoidal choroidal vasculopathy. Invest. Ophthalmol. Vis. Sci. 49, 2613–2619 (2008).
Huang, L., Li, Y., Guo, S., Sun, Y., Zhang, C., Bai, Y. et al. Different hereditary contribution of the CFH gene between polypoidal choroidal vasculopathy and age-related macular degeneration in Chinese Han people. Invest. Ophthalmol. Vis. Sci. 55, 2534–2538 (2014).
Huang, L., Zhang, H., Cheng, C. Y., Wen, F., Tam, P. O., Zhao, P. et al. A missense variant in FGD6 confers increased risk of polypoidal choroidal vasculopathy. Nat. Genet. 48, 640–647 (2016).
Cheng, C. Y., Yamashiro, K., Chen, L. J., Ahn, J., Huang, L., Huang, L. et al. New loci and coding variants confer risk for age-related macular degeneration in East Asians. Nat. Commun. 6, 6063 (2015).
Japanese Study Group of Polypoidal Choroidal, V Criteria for diagnosis of polypoidal choroidal vasculopathy. Nippon Ganka Gakkai Zasshi 109, 417–427 (2005).
Delaneau, O., Marchini, J. & Zagury, J. F. A linear complexity phasing method for thousands of genomes. Nat. Methods 9, 179–181 (2011).
Genomes Project, C. Genomes Project, C., Abecasis, G. R. Genomes Project, C., Altshuler, D. Genomes Project, C., Auton, A. Genomes Project, C., Brooks, L. D. Genomes Project, C., Durbin, R. M. et al. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010).
Howie, B. N., Donnelly, P. & Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 5, e1000529 (2009).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Rivas, M. A., Pirinen, M., Conrad, D. F., Lek, M., Tsang, E. K., Karczewski, K. J. et al. Human genomics. Effect of predicted protein-truncating genetic variants on the human transcriptome. Science 348, 666–669 (2015).
Liu, J. Z., McRae, A. F., Nyholt, D. R., Medland, S. E., Wray, N. R., Brown, K. M. et al. A versatile gene-based test for genome-wide association studies. Am. J. Hum. Genet. 87, 139–145 (2010).
Bulik-Sullivan, B., Finucane, H. K., Anttila, V., Gusev, A., Day, F. R., Loh, P. R. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241 (2015).
Ma, L., Li, Z., Liu, K., Rong, S. S., Brelen, M. E., Young, A. L. et al. Association of genetic variants with polypoidal choroidal vasculopathy: a systematic review and updated meta-analysis. Ophthalmology 122, 1854–1865 (2015).
Yanagisawa, S., Kondo, N., Miki, A., Matsumiya, W., Kusuhara, S., Tsukahara, Y. et al. Difference between age-related macular degeneration and polypoidal choroidal vasculopathy in the hereditary contribution of the A69S variant of the age-related maculopathy susceptibility 2 gene (ARMS2). Mol. Vis. 17, 3574–3582 (2011).
Miki, A., Honda, S., Kondo, N. & Negi, A. The association of age-related maculopathy susceptibility 2 (ARMS2) and complement factor H (CFH) variants with two angiographic subtypes of polypoidal choroidal vasculopathy. Ophthal. Genet. 34, 146–150 (2013).
Yanagisawa, S., Sakurada, Y., Miki, A., Matsumiya, W., Imoto, I. & Honda, S. The association of elastin gene variants with two angiographic subtypes of polypoidal choroidal vasculopathy. PLoS ONE 10, e0120643 (2015).
Amin, E. M., Oltean, S., Hua, J., Gammons, M. V., Hamdollah-Zadeh, M., Welsh, G. I. et al. WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell 20, 768–780 (2011).
Chen, H., Liu, K., Chen, L. J., Hou, P., Chen, W. & Pang, C. P. Genetic associations in polypoidal choroidal vasculopathy: a systematic review and meta-analysis. Mol. Vis. 18, 816–829 (2012).
Mittlbock, M. & Schemper, M. Explained variation for logistic regression. Stat. Med. 15, 1987–1997 (1996).
Acknowledgements
We gratefully thank all the participants or volunteers who participated in the studies. This study was supported by the National Medical Research Council (NMRC grants 0796/2003, IRG07nov013, IRG09nov014, NMRC 1176/2008, NIG/1003/2009, STaR/0003/2008, CG/SERI/2010 and CSA/033/2012) and Biomedical Research Council (BMRC 08/1/35/19/550, 09/1/35/19/616 and 10/1/35/19/671) in Singapore; Direct Grants of the Chinese University of Hong Kong (4054119, CPP and 2015.1.045, LJC, Hong Kong); BrightFocus Foundation (M2011068), USA; the Seoul National University Bundang Hospital Research Grant Fund (grant number 03-2009-008) and National Research Foundation of Korea (NRF-2009-0072603, NRF-2012R1A1A2008943 and NRF-2014R1A2A1A09005824) grants funded by the Ministry of Education, Science and Technology, Korea; and grants-in-aid for scientific research (number 24249082) from the Japan Society for the Promotion of Science,Tokyo, Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Fan, Q., Cheung, C., Chen, L. et al. Shared genetic variants for polypoidal choroidal vasculopathy and typical neovascular age-related macular degeneration in East Asians. J Hum Genet 62, 1049–1055 (2017). https://doi.org/10.1038/jhg.2017.83
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2017.83
This article is cited by
-
Macular neovascularization and polypoidal choroidal vasculopathy: phenotypic variations, pathogenic mechanisms and implications in management
Eye (2024)
-
The hypothetical molecular mechanism of the ethnic variations in the manifestation of age-related macular degeneration; focuses on the functions of the most significant susceptibility genes
Graefe's Archive for Clinical and Experimental Ophthalmology (2024)
-
Contribution of common and rare variants to Asian neovascular age-related macular degeneration subtypes
Nature Communications (2023)
-
Prevalence of and factors associated with dilated choroidal vessels beneath the retinal pigment epithelium among the Japanese
Scientific Reports (2021)
-
The association of polypoidal choroidal vasculopathy clinical phenotypes with previously reported genetic markers
Graefe's Archive for Clinical and Experimental Ophthalmology (2020)
