
Abstract
Familial episodic pain is a rare autosomal-dominant disorder characterized by recurrent attacks of pain. The pathogenesis of familial episodic pain is not very clear so far. Essential tremor is the most common movement disorder, but the identification of essential tremor genes has remained elusive. We studied a four-generation Chinese family with early-onset familial episodic pain and adult onset familial essential tremor. All essential tremor diagnoses were confirmed based on a review of the questionnaires, videotaped neurological examinations and was then reconfirmed by a senior neurologist specializing in movement disorders using published criteria. SCN11A analysis was performed by whole-exome sequencing or Sanger sequencing. We confirmed the presence of the SCN11A (c.673C>T) mutation in family members with episodic pain and essential tremor. We identified a missense mutation of p.Arg225Cys in SCN11A in a four-generation Chinese family with early-onset familial episodic pain and adult onset familial essential tremor syndrome. This may belong to a rare hereditary syndrome that has not been reported up to now. For the first time, we associated the genetic variability of SCN11A with the development of essential tremor, and further confirmed essential tremor is one of the neurological channelopathies.
Similar content being viewed by others

Introduction
Familial episodic pain is a rare autosomal-dominant disorder characterized by recurrent attacks of pain. The intense pain appeared in infancy was highest in childhood and gradually reduced with the increase of age. The pain is often localized to the distal limbs, especially joints. Episodic pain is triggered by conditions of fatigue, strenuous exercise and cold. The pain was relieved by taking anti-inflammatory analgesic drugs (paracetamol tablets and Fenbid capsules). Until now, there are two genes (TRPA1 and SCN11A), which have been linked to this pain syndrome. The difference is that TRPA1 mutation brought about familial episodic pain localizing mainly the upper body,1 however the pain resulted in SCN11A mutations principally affecting the distal lower extremities.2, 3 The pathogenesis of familial episodic pain is not very clear so far. The research of this disease is still in its initial stage.
Essential tremor (ET) is one of the most frequent neurological disorders and the most common movement disorder among adults whose incidence increases with age. Its core motor symptom is a bilateral postural and kinetic tremor of the hands and arms, some patients with ET also develop other motor and non-motor manifestations including Parkinsonism, myoclonus, dystonia, cerebellar dysfunction, sensory abnormalities, sleep disorders, and cognitive and psychiatric features.4 However, the etiology of ET is still unknown, the genetic cause of ET remains elusive. There is growing evidence that ET may not be a single disease but rather a family of diseases.5 Previously, genome-wide linkage studies in families mapped three loci for ET, hereditary essential tremor-1 (ETM1), ETM2 and ETM3, but no causal mutation has been replicated in candidate genes within these loci.6 Genetic variants within the LINGO1 (MIM #609791), DRD3 (MIM #126451) and HS1-BP3 (MIM#609359) genes have been reported to associate with ET,7, 8 but these associations have not been consistently replicated. Polymorphisms within the SLC1A2 (MIM#600300) gene have also been reported to increase susceptibility to ET,9 and the mutations in FUS (MIM #137070), HTRA2 (MIM #606441) and SORT1 (MIM #602458) genes have been reported to cause ET with whole-exome sequencing analyses.10, 11, 12 Recently, missense mutations in TENM4 from a Spanish population have been reported to cause ET.13 More recently, SCN4A pore mutation has been reported to contribute to autosomal-dominant ET.14 This foundation suggested ET maybe one of the neurological channelopathies. Up to now, no human heritable disorders of ET have been linked to mutations in SCN11A channels.
The voltage-sensitive Na+ channel is a transmembrane protein essential for the generation of action potentials in excitable cells.15 The family of mammalian NaV channels contains nine members (NaV1.1–1.9) encoded by the genes SCN1A–SCN5A and SCN8A–SCN11A.16 Sodium channels in mammals include an α subunit that encodes the core protein of the channel and auxiliary β subunits that modify the channel function.17 Every α subunit of sodium channels includes four domains (DI–DIV) and each domain consists of six transmembrane segments (S1–S6).15 In these nine subtypes (Nav1.1–Nav1.9) of sodium channels, three subtypes (Nav1.7, Nav1.8 and Nav1.9) are strongly expressed in sensory neurons, and have been linked with human pain disorders. Nav1.4 has been associated with autosomal-dominant ET recently.14
SCN11A, which encodes Nav1.9, is a tetrodotoxin-resistant channel preferentially expressed in nociceptors of the peripheral nervous system. Nav1.9 channels exhibits unique biophysical characteristics, possesses hyperpolarized voltage-dependent activation close to the resting membrane potential, with slow activation and inactivation leading to a persistent current.18 The amplitude of persistent sodium currents-mediated NaV1.9 can be increased by inflammatory mediators in a G-protein-coupled pathway.19 It has been suggested that NaV1.9 is likely capable of regulating resting neuronal excitability, and acts as a threshold channel to modulate neuronal excitability by prolonging the depolarizing effects of sub-threshold stimuli.20, 21 There have been relatively few reports about SCN11A mutations and related diseases. Recently SCN11A mutation was reported to have a major role in some peripheral pain syndromes. A novel missense SCN11A mutation (p.L811P) was found in a German family with loss-of-pain sensation.22 Nevertheless, two types of gain-of-function SCN11A mutations (R225C and A808G) were identified by genome-wide linkage analysis combined with whole-exome sequencing in two large multigenerational Chinese families with autosomal-dominant episodic chronic pain.2 More recently, a NaV1.9 mutation (p.V1184A) was reported that causes early-onset cold-aggravated familial episodic pain,23 and two missense SCN11A variants (R222H and R222S) were identified in six unrelated multigenerational Japanese families with episodic pain syndrome.3
Up till now, it has been unclear whether mutations in SCN11A are associated with human genetic ET. So further studies are essential to explore phenotype–genotype correlations in Nav1.9 channelopathy.
In this study, we here describe a NaV1.9 mutation (p.Arg225Cys) that causes child onset familial episodic pain and adult onset familial ET. We have successfully identified a novel mutation of SCN11A associated with ET in a Chinese family with eight affected individuals.
Materials and methods
Subjects
A 5-year-old boy from China, Beijing (Chinese Han) was hospitalized because of recurrent episodic extremities pain attacks associated with sweating from infancy. Ten members in his four-generation family had similar symptoms. Among the 10 affected members, 8 members are adults, 2 members are children. All the affected members have ET after they entered adulthood. After the informed consent was obtained from all participants, peripheral blood samples were obtained from eight affected and five unaffected family members, and used for isolation of genomic DNA. This study was approved by the ethics committee of Xuanwu Hospital Capital Medical University.
SCN11A mutation analysis
To appraise the pathogenic mutations, peripheral blood sample from the proband of the family was sent to Kangso medical inspection (Beijing, China) for whole-exome sequencing. Experimental procedures included the following five steps. Step 1: Genomic DNA was extracted from blood samples by the FlexiGene DNA Kit. Step2: Genomic DNA was randomly fragmented by the Diagenode Ultrasonic crusher and the size of the library fragments was distributed mainly between 100 and 500 bp, and ligated by adapters, hybridized, amplified and purified with a Agilent SureSelect XT Human All Exon V6 library. Step 3: Captured DNA products were subjected to Agilent 2100 Bioanalyzer (American Agilent company, Santa Clara, CA, USA) to estimate the magnitude of enrichment. After quality control, sequencing was performed on the Illumina sequencing machine. Step 4: Clean reads were analysed with software samtools, picard and BWA for single-nucleotide polymorphism detection, insertion detection and deletion detection, respectively. The called single-nucleotide polymorphisms and indels were annotated by software PolyPhen-2.2.2, ANNOVAR and HGMD databases. db single-nucleotide polymorphism was used to filter known variants. Protein functions of missense single-nucleotide polymorphism were predicted with 1000 Genomes Project Database. Step 5: The suspicious mutation was further confirmed by Sanger sequencing in all affected individuals and five unaffected members as a control.
Results
Clinic features of a Chinese family with familial episodic pain and essential tremor
The four-generation pedigree was shown in Figure 1. A 5-year-old proband (IV-12) visited our hospital in August 2015 when he experienced recurrent episodic extremities pain attacks associated with sweating. He had similar pain attacks for the past 3 years, with a frequency once per 3–5 days. He started recurrent episodes of unexplained crying at 1 year of age, which was recognized to be the result of limb pain. The severe pain was localized principally to the distal lower extremities and occasionally in the upper limb, especially in the arthrosis of legs and arms. The degree of pain is intense, unbearable. The pain appeared before going to sleep at night and disappeared after dawn. In a strange way, the episodic pain attacked at regular intervals (1 h) for a total of 5–8 recurrences for one cycle, and lasted for 15–20 min every time. The pain was induced with fatigue, cold, sleep deprivation and strenuous exercise. The pain was relieved by taking anti-inflammatory analgesic drugs, and was alleviated by a hot compress. The neurological examination revealed no obvious abnormalities. Brain magnetic resonance imaging was normal. Electroencephalogram recording revealed normal activity during pain attack. During hospitalization, the patient had two pain attacks with 3–4 days intervals.

Pedigree structures in a Chinese family with episodic pain and essential tremor. Individuals with episodic pain are indicated by black solid squares (males) or black solid circles (females). Individuals with episodic pain and essential tremor are indicated by black and green solid squares (males) or black and green solid circles (females). Unaffected individuals are indicated by open symbols. Deceased individuals are indicated by slashes (/). The proband is indicated by an arrow.
On further inquiry about the family history, we found that nine other members in his four-generation family had similar symptoms and showed slightly variable expressivity. Then, affected individuals in the family were evaluated. All visited individuals were numbered (Figure 1) and their DNA samples were obtained. All affected individuals from the family have most similar clinical symptom characteristics according to their descriptions. First, sharp pain was usually at the distal lower extremities and occasionally at arms, especially at knee joints. Second, severe pain appeared at night and the degree of pain is intense, unbearable. Third, episodic pain attacked at regular intervals (1 h), and repeated once every 2–14 days for a total of 4–8 recurrences for one cycle. Fourth, the pain was induced with fatigue, cold and strong movement. The pain was relieved by taking anti-inflammatory analgesic drugs (paracetamol tablets and Fenbid capsules), and was alleviated by a hot compress. Fifth, intense pain usually was along with sweating. At last but not least, intense pain appeared in infancy, it was highest in childhood and gradually reduced with the increase of age, from the age of 18 has occurred only 1–2 times per year, and disappeared gradually during adolescence. The clinical symptoms and signs of all affected individuals were detailed in Table 1.
Interestingly enough, except for episodic pain of this family, all affected individuals of the family showed ET when stepped into adulthood. Afterwards, all ET diagnoses were confirmed based on a review of the questionnaires, videotaped neurological examinations and electromyogram. An in-person evaluation was conducted in the individuals suffered from tremor, this contained a series of questionnaires about their tremor and their use of medications. The videotaped neurological examination comprised a detailed assessment of postural, kinetic, intention and rest tremors in the limbs, as well as dystonia and other movement disorders. After review of the history, videotaped examinations and electromyogram, the diagnosis of ET was then reconfirmed by a senior neurologist specializing in movement disorders using published criteria.24 All affected individuals from the family have most common clinical symptom features. First, the ET was just localized to the double upper limbs, especially at both hands and arms, without voice, head or other parts of tremors. Second, the ET appeared after pain disappeared, all affected cases were adults aged from 30 to 41 years old when tremor appeared. Third, all affected individuals initially had both postural and action tremor, and later developed resting tremor gradually aggravated with age. Fourth, the ET frequency of all affected individuals from the family was 4–8 Hz, which was recorded by electromyogram. The ET wasn’t noticeable in early (amplitude <1 cm), and obviously aggravated in later period (amplitude 1–3 cm). The tremor frequency decreased and amplitude increased with age. Fifth, no other signs of motor impairment suggestive of Parkinsonism, myoclonus, dystonia and ataxia were identified. Sixth, all the affected individuals had no symptoms of sensory disturbance like local numbness, burning pain, pricking pain and movement disorder such as muscular atrophy suggestive of peripheral neuropathy. The neurological examination revealed that the deep tendon reflexes in the upper extremities of all affected individuals were preserved. Electromyogram indicated no nerve lesion in all the affected individuals. The clinical symptoms and signs of all individuals suffered from tremor were showed in Table 2.
Identification of SCN11A as the disease-causing gene
To identify the pathogenic mutations, peripheral blood sample from the proband of the family was sent to Kangso medical inspection (Beijing, China) for whole-exome sequencing. A variant (c.673C>T) in SCN11A (RefSeq accession number NM_014139.2) was found, which was first reported to cause familial episodic pain in 2013. To verify cosegregation of the SCN11A mutations in family members with episodic pain and ET, we performed Sanger sequencing in the other seven affected (II-1, II-2, III-6, III-7, III-9, IV-11, IV-13) and five unaffected family members (II-3, III-4, III-5, III-8, IV-10) with available DNA (Figure 1). We confirmed the presence of the SCN11A (c.673C>T) mutation in family members with episodic pain and ET. All unaffected family members did not carry the SCN11A (c.673C>T) mutation.
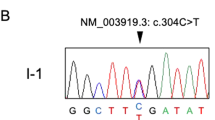
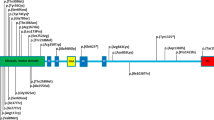
So a variant (c.673C>T) in SCN11A was found, which was corresponding to the pain and ET phenotype in the affected persons of the family. This is the first time to identify SCN11A mutation associate with ET. Sanger sequencing results were shown as in Figure 2. This mutation results in an Arg to Cys substitution at amino acid residue 225 (p.Arg225Cys) in the transmembrane segment S4–DI region of the Nav1.9 channel protein (Figure 2). The voltage-gated sodium channel NaV1.9 is preferentially expressed in peripheral neurons nociceptors and has been shown in rodent models to have a major role in inflammatory and neuropathic pain. Recently, mutations in SCN11A have been identified in individuals with rare genetic episodic pain diseases and in larger populations of people with painful peripheral neuropathy,2, 3, 25 whereas another study described a mutation in SCN11A was associated with a congenital inability to experience pain.22

SCN11A Mutation from the family with episodic pain and essential tremor. (a) DNA sequence chromatograms showing the mutation in SCN11A identified in our study family. (b) Schematic structure diagram of the Nav1.9 protein with pain- and tremor-associated mutation.
No pathogenic mutations were found in other genes that cause pain like TRPA1, SCN9A and SCN10A. As the family showed ET when stepped into adulthood, we pay special attention to ET-associated gene in the proband, but no pathogenic mutations that associate with ET like ETM1, ETM2, ETM3, LINGO1, DRD3, HS1-BP3, SLC1A2, FUS, HTRA2, SORT1,TENM4 and SCN4A were found.
Discussion
Here we described a Chinese four-generation family with familiar episodic pain in childhood and ET in adulthood. Ten members in the family were affected. All the affected individuals experienced recurrent episodic extremities pain attacks in childhood. The intense episodic pain appeared in infancy, gradually reduced with the increase of age, and subsided spontaneously when stepped into adulthood. All episodic pain-affected individuals showed ET when they entered adulthood. The ET appeared after pain disappeared without accompanying Parkinsonism, myoclonus, dystonia and ataxia. This may belong to a rare hereditary syndrome, and has not been reported up to now. This syndrome is the first time to make a connection between episodic pain and ET. Thus, further studies are warranted to elucidate the mechanisms of such an age-dependent syndrome.
In the present study, we identified SCN11A R225C mutation in a Chinese family with familial episodic pain in childhood and ET in adulthood. There are five SCN11A mutations, which has been reported caused familiar episodic pain. Zhang et al.2 first reported two large multigenerational Chinese families with autosomal-dominant episodic chronic pain and two types of gain-of-function SCN11A mutations (R225C and A808G) were identified by genome-wide linkage analysis combined with whole-exome sequencing. Recent studies in humans have reported that other three amino acid substitutions of Nav1.9 (V1184A, R222H and R222S) are relevant to familiar episodic pain disorder.3, 23 Therefore, SCN11A R225C may be a hot mutation result in familiar episodic pain, especially in Chinese people. R225C located in the transmembrane segment S4 at DI region of the Nav1.9 channel protein, R222H and R222S also located at DI region,3 so DI region may be the most important functional area of the Nav1.9 channel protein, and further studies are needed about DI region of the Nav1.9 channel protein.
The mutation of SCN11A R225C in this study becomes interesting because direct link of Nav1.9 to human ET is the first time to report. In our study, we found a variant (c.673C>T) in SCN11A, which was corresponding to ET phenotype in the affected persons of the family using whole-exome sequencing. ET is one of the most common movement disorders with a poorly understood etiology, the genetic architecture of ET remains unknown. Combined with previous research, although these associations have not been consistently replicated, possible candidate genes for ET contained ETM1, ETM2, ETM3, LINGO1, DRD3, HS1-BP3, SLC1A2, FUS, HTRA2,SORT1 and TENM4 have been reported.6, 7, 8, 9, 10, 11, 12, 13, 26 We pay special attention to ET-associated gene above-mentioned in the proband, but no pathogenic mutations were found. A recent study has shown that autosomal-dominant ET caused by a gain-of-function mutation in SCN4A is correlated with natural change of Nav1.4 channel.14 This suggested ET may associate with neurological channelopathies. In our study, we associate the SCN11A mutation with the development of ET and further confirm ET is one of the neurological channelopathies.
The SCN11A R225C mutation has been reported caused familial episodic pain in a Chinese family without ET, what is the reason for this difference? We speculate the reason may be that ET can be caused by an autosomal-dominant gene with low penetrance. Despite reports of several ET families with typical autosomal-dominant inheritance,27, 28, 29, 30, 31 the frequency of affected first-degree relatives reported in some studies is ~23%,32, 33, 34 much lower than the 50% expected for autosomal-dominant inheritance with complete penetrance and slightly lower than the 25% expected for autosomal-recessive inheritance. Therefore, ET has the possibility of a multifactorial inheritance pattern, environmental factors should be also taken into account. In an attempt to answer the presence of phenocopies in families with ET and to explain the non-Mendelian features in the genetics of this disease, Zimprich35 proposed the hypothesis of a possible role of epigenetic factors in the inheritance of ET including heritable changes in gene expression or cellular phenotype caused by mechanisms other than changes in the nucleotide sequence or functionally relevant modifications to the genome that do not involve a change in the underlying DNA sequence.34 There is another possibility that the actual mutation pathogenic for ET might be located in the vicinity with close linkage of SCN11A, not the SCN11A mutation itself. SCN11A is located in 3p22.2. RNU6-1227P, WDR48, TTC21A, GORASP1 are located in physical proximity to SCN11A, which we found in NCBI gene bank, there are no pathogenic mutations in these genes using whole-exome sequencing. And the other genes such as MLH1, CTNNB1, TGFBR2, SCN5A, which are also located in 3p22.2 have no pathogenic mutations too.
The mutation (c.673C>T) in SCN11A results in an Arg to Cys substitution at amino acid residue 225 (p.Arg225Cys) in the transmembrane segment S4 at DI region of the Nav1.9 channel protein. Then, how the SCN11A mutation brings about episodic pain and ET? Zhang et al.2 had a functional analysis of the SCN11A mutation (c.673C>T). They expressed the SCN11A mutant in mouse dorsal root ganglion neurons and showed that the mutation enhanced the channel’s electrical activities and induced hyperexcitablity of dorsal root ganglion neurons using conventional whole-cell patch-clamp recording.2 It has been proposed that the Nav1.9 channel relates to the modulation of nociceptor membrane potential and contributes to establish the membrane voltage, thus that may in turn influence gating of other Nav channels present in neurons and overall neuronal excitability.20, 36 In general, the higher electrical activities of mutant Nav1.9 channel might significantly alter the membrane potentials and lead to the opening of other Nav channels that cause neurons to be hyperexcitable, which contributed to the familial episodic pain and ET.
As described in previous reports, the pain is also alleviated in patients from the Chinese family by oral administration of anti-inflammatory drugs. The pain-alleviating effects of the anti-inflammatory therapy correlate with an observed upregulation of NaV1.9 under inflammatory conditions37 and emphasize a role of NaV1.9 in mediating inflammatory pain. As the ET did not seriously interfere with life, patients from the Chinese family did not take any medication to treat. In our study, we identified a missense mutation of p.Arg225Cys in SCN11A in a Chinese family with familial episodic pain in childhood and ET in adulthood syndrome. This syndrome suggests that there may be the same pathogenesis of tremor and pain. This may explain why some medicine not only to be effective on pain, but also to improve movement disorders such as Parkinson's disease, tremor. Interestingly, our above analysis demonstrated that the Nav1.9 channel would be an appealing target for the development of specific drugs to relieve tremor.
In summary, a missense mutation of p.Arg225Cys in SCN11A was identified in a four-generation Chinese family with familial episodic pain in childhood and ET in adulthood syndrome. This may belong to a rare hereditary syndrome that has not been reported up to now. This syndrome is the first time to make a connection between episodic pain and ET. It is the first time to report that a SCN11A mutation contribute to ET, and further confirm ET is one of the neurological channelopathies.
References
Kremeyer, B., Lopera, F., Cox, J. J., Momin, A., Rugiero, F., Marsh, S. et al. A gain-of-function mutation in TRPA1 causes familial episodic pain syndrome. Neuron 66, 671–680 (2010).
Zhang, X. Y., Wen, J., Yang, W., Wang, C., Gao, L., Zheng, L. H. et al. Gain-of-function mutations in SCN11A cause familial episodic pain. Am. J. Hum. Genet. 93, 957–966 (2013).
Okuda, H., Noguchi, A., Kobayashi, H., Kondo, D., Harada, K. H., Youssefian, S. et al. Infantile pain episodes associated with novel Nav1.9 mutations in familial episodic pain syndrome in Japanese Families. PLoS ONE 11, e0154827 (2016).
Louis, E. D. Essential tremor: evolving clinicopathological concepts in an era of intensive post-mortem enquiry. Lancet Neurol. 9, 613–622 (2010).
Benito-Leon, J. Essential tremor: a neurodegenerative disease? Tremor Other Hyperkinet. Mov. 4, 1–9 (2014).
Jasinska-Mygaa, B. & Widerb, C. Genetics of essential tremor. Parkinsonism Relat. Disord. 18S1, S138–S139 (2012).
Kuhlenbaumer, G., Hopfner, F. & Deuschl, G. Genetics of essential tremor: meta-analysis and review. Neurology 82, 1000–1007 (2014).
Zimprich, A. Genetics of Parkinson’s disease and essential tremor. Curr. Opin. Neurol. 24, 318–323 (2011).
Thier, S., Lorenz, D., Nothnagel, M., Poremba, C., Papengut, F., Appenzeller, S. et al. Polymorphisms in the glial glutamate transporter SLC1A2 are associated with essential tremor. Neurology 79, 243–248 (2012).
Merner, N. D., Girard, S. L., Catoire, H., Bourassa, C. V., Belzil, V. V., Riviere, J. B. et al. Exome sequencing identifies FUS mutations as a cause of essential tremor. Am. J. Hum. Genet. 91, 313–319 (2012).
Unal Gulsuner, H., Gulsuner, S., Mercan, F. N., Onat, O. E., Walsh, T., Shahin, H. et al. Mitochondrial serine protease HTRA2 p.G399S in a kindred with essential tremor and Parkinson disease. Proc. Natl Acad. Sci. USA 111, 18285–18290 (2014).
Sánchez, E., Bergareche, A., Krebs, C. E., Gorostidi, A., Makarov, V., Ruiz-Martinez, J. et al. SORT1 mutation resulting in sortilin deficiency and p75NTR upregulation in a family with essential tremor. ASN Neuro 7, 1759091415598290 (2015).
Hor, H., Francescatto, L., Bartesaghi, L., Ortega-Cubero, S., Kousi, M., Lorenzo-Betancor, O. et al. Missense mutations in TENM4, a regulator of axon guidance and central myelination, cause essential tremor. Hum. Mol. Genet. 24, 5677–5686 (2015).
Bergareche, A., Bednarz, M., Sánchez, E., Krebs, C. E., Ruiz-Martinez, J., De La Riva, P. et al. SCN4A pore mutation pathogenetically contributes to autosomal dominant essential tremor and may increase susceptibility to epilepsy. Hum. Mol. Genet. 24, 7111–7120 (2015).
Ogata, N. & Ohishi, Y. Molecular diversity of structure and function of the voltage-gated Nat channels. Jpn. J. Pharmacol. 88, 365–377 (2002).
Goldin, A. L., Barchi, R. L., Caldwell, J. H., Hofmann, F. & Howe, J. R. Nomenclature of voltage-gated sodium channels. Neuron 28, 365–368 (2000).
Isom, L. L., De-Jongh, K. S., Patton, D. E., Reber, B. F., Offord, J., Charbonneau, H. et al. Primary structure and functional expression of the β1 subunit of the rat brain sodium channel. Science 256, 839–842 (1992).
Dib-Hajj, S. D., Black, J. A. & Waxman, S. G. NaV1.9: a sodium channel linked to human pain. Nat. Rev. Neurosci. 16, 511–519 (2015).
Maingret, F., Coste, B., Padilla, F., Clerc, N., Crest, M., Korogod, S. M. et al. Inflammatory mediators increase Nav1.9 current and excitability in nociceptors through a coincident detection mechanism. J. Gen. Physiol. 131, 211–225 (2008).
Herzog, R. I., Cummins, T. R. & Waxman, S. G. Persistent TTX-resistant Na+ current affects resting potential and response to depolarization in simulated spinal sensory neurons. J. Neurophysiol. 86, 1351–1364 (2001).
Baker, M. D., Chandra, S. Y., Ding, Y., Waxman, S. G. & Wood, J. N. GTP-induced tetrodotoxin-resistant Na+ current regulates excitability in mouse and rat small diameter sensory neurones. J. Physiol. 548, 373–382 (2003).
Leipold, E., Liebmann, L., Korenke, G. C., Heinrich, T., Giesselmann, S., Baets, J. et al. A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nat. Genet. 45, 1399–1404 (2013).
Leipold, E., Hanson-Kahn, A., Frick, M., Gong, P., Bernstein, J. A., Voigt, M. et al. Cold-aggravated pain in humans caused by a hyperactive NaV1.9 channel mutant. Nat. Commun. 6, 10049 (2015).
Louis, E. D., Ford, B. & Bismuth, B. Reliability between two observers using a protocol for diagnosing essential tremor. Mov. Disord. 13, 287–293 (1998).
Huang, J., Han, C., Estacion, M., Vasylyev, D., Hoeijmakers, J. & Gerrits, M. Gain-of-function mutations in sodium channel Nav1.9 in painful neuropathy. Brain 137, 1627–1642 (2014).
Murni Tio, A. & Eng-King, T. Genetics of essential tremor. Parkinsonism Relat. Disord. 22, S176–S178 (2016).
Bain, P. G., Findley, L. J. & Thompson, P. D. A study of hereditary essential tremor. Brain 117, 805–824 (1994).
Louis, E. D. & Ottman, R. How familial is familial tremor? The genetic epidemiology of essential tremor. Neurology 46, 1200–1205 (1996).
Jankovic, J., Beach, J., Pandolfo, M. & Patel, P. I. Familial essential tremor in 4 kindreds. Prospects for genetic mapping. Arch. Neurol. 54, 289–294 (1997).
Kovach, M. J., Ruiz, J. & Kimonis, K. Genetic heterogeneity in autosomal dominant essential tremor. Genet. Med. 3, 197–199 (2001).
Pasini, E., Busolin, G., Nobile, C. & Michelucci, R. Autosomal dominant essential tremor: a novel family with anticipation. Neurol. Sci. 34, 761–763 (2013).
Jankovic, J., Beach, J., Schwartz, K. & Contant, C. Tremor and longevity in relatives of patients with Parkinson’s disease, essential tremor, and control subjects. Neurology 45, 645–648 (1995).
Louis, E. D., Ford, B., Frucht, S., Barnes, L. F., X-Tang, M. & Ottman, R. Risk of tremor and impairment from tremor in relatives of patients with essential tremor: a community-based family study. Ann. Neurol. 49, 761–769 (2001).
Jiménez-Jiménez, F. J., Alonso-Navarro, H., García-Martín, E., Lorenzo-Betancor, O., Pastor, P. & Agúndez, J. A. Update on genetics of essential tremor. Acta Neurol. Scand. 128, 359–371 (2013).
Zimprich, A. Phenocopies in families with essential tremor and restless legs syndrome challenge Mendelian laws. Epigenetics might provide answers. Parkinsonism Relat. Disord. 18, 711–716 (2012).
Dib-Hajj, S. D., Cummins, T. R., Black, J. A. & Waxman, S. G. Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 33, 325–347 (2010).
Maingret, F., Coste, B., Padilla, F., Clerc, N., Crest, M., Korogod, S. M. et al. Inflammatory mediators increase Nav1.9 current and excitability in nociceptors through a coincident detection mechanism. J. Gen. Physiol. 131, 211–225 (2008).
Acknowledgements
We thank all subjects and families who participated in the study. This work was supported by the National Natural Science Foundation of China, grant number: 81271494.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Leng, XR., Qi, XH., Zhou, YT. et al. Gain-of-function mutation p.Arg225Cys in SCN11A causes familial episodic pain and contributes to essential tremor. J Hum Genet 62, 641–646 (2017). https://doi.org/10.1038/jhg.2017.21
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2017.21
This article is cited by
-
Association Analysis of 27 Single Nucleotide Polymorphisms in a Chinese Population with Essential Tremor
Journal of Molecular Neuroscience (2023)
-
Pathological changes of the sural nerve in patients with familial episodic pain syndrome
Neurological Sciences (2022)
-
Gene Expression Analysis of Laser-Captured Purkinje Cells in the Essential Tremor Cerebellum
The Cerebellum (2022)
-
Essential tremor
Nature Reviews Disease Primers (2021)
-
Current Opinions and Consensus for Studying Tremor in Animal Models
The Cerebellum (2019)
