
Abstract
It is considered that more than 15 depths of coverage are necessary for next-generation sequencing (NGS) data to obtain reliable complete nucleotide sequences of the mitogenome. However, it is difficult to satisfy this requirement for all nucleotide positions because of problems obtaining a uniform depth of coverage for poorly preserved materials. Thus, we propose an imputation approach that allows a complete mitogenome sequence to be deduced from low-depth-coverage NGS data. We used different types of mitogenome data files as panels for imputation: a worldwide panel comprising all the major haplogroups, a worldwide panel comprising sequences belonging to the estimated haplogroup alone, a panel comprising sequences from the population most closely related to an individual under investigation, and a panel comprising sequences belonging to the estimated haplogroup from the population most closely related to an individual under investigation. The number of missing nucleotides was drastically reduced in all the panels, but the contents obtained by imputation were quite different among the panels. The efficiency of the imputation method differed according to the panels used. The missing nucleotides were most credibly imputed using sequences of the estimated haplogroup from the population most closely related to the individual under investigation as a panel.
Similar content being viewed by others

Introduction
Human population studies rely greatly on mitochondrial DNA (for example, Smith1). Recently, complete mitogenome sequences have been accumulated for various contemporary human populations worldwide, and detectable differences can be observed even among closely related populations. Recombination does not fundamentally occur because of the haploidy of the mitogenome. Therefore, population histories have been considered based on various analyses such as reconstructions of demographic history using Bayesian Skyline Plots and estimating the time to the most recent common ancestor using the Markov chain Monte Carlo method (for example, Mizuno et al.2; Gojobori et al.3). In addition, ancient mitogenome sequences are expected to provide direct evidence of what happened in our past, such as migration, demography, and the relationships among populations. However, most human remains have been excavated from temperate and subtropical regions where the buried remains have often undergone microbial attack, and the conditions for DNA preservation are generally poor.4, 5 To overcome this problem, we previously proposed a unified method in which emulsion PCR was coupled with target enrichment, followed by next-generation sequencing (NGS).6 This unified method facilitates a more efficient determination of non-duplicated target sequences than shotgun NGS, and we successfully achieved deep and reliable DNA sequencing of the ancient mitogenome, even using poorly preserved archeological samples. However, this problem is still challenging. Environmental conditions such as humidity, temperature, salinity, pH and microbial attack strongly influence the degree of DNA preservation.7 Thus, it is considered that more than 15 depths of coverage NGS data are necessary to obtain reliable complete nucleotide sequences of haploid-like genomes such as the mitogenome and Y chromosome.8 However, it is difficult to satisfy this requirement in all nucleotide positions because of problems obtaining a uniform depth of coverage, particularly for poorly preserved materials such as human remains and archeological samples. Therefore, the mitogenome sequences obtained are often fragmentary. To fill in the missing sequences (nucleotides) in the mitogenome, we propose an imputation approach for estimating missing mitogenome sequences. Imputation has been used widely in genome-wide association studies to predict the genotypes at single-nucleotide polymorphism (SNP) sites.9, 10, 11 The advantage of the imputation approach is that it is possible to use the maximum number of samples with the longest sequence in various analyses for population genetic studies. We applied the imputation approach to mitogenome data from a 1500-year-old human remains excavated from the Moon Pyramid at Teotihuacan, Mexico, where the depth of coverage was very low due to poor preservation of the DNA.
Materials and methods
Imputation
Missing nucleotides were inferred using a k-nearest neighbor (KNN)-based algorithm.12 On the basis of 22,638 worldwide mitogenome sequences, we prepared four types of mitogenome panel. Panel 1 as a worldwide panel comprising 292 mitogenome sequences of 29 haplogroups (L, L3, M, C, E, G, Q, Z, D, N, A, I, O, S, W, X, Y, R, B, F, J, P, T, R0, HV, H, V, U and K); Panel 2 as a worldwide haplogroup A panel comprising 731 mitogenome sequences; Panel 3 as an indigenous American panel comprising 390 mitogenome sequences; Panel 4 as an indigenous American haplogroup A panel comprising 175 mitogenome sequences. This haplogroup nomenclature is based on PhyloTree Build 17 (http://www.phylotree.org/).13 In addition to complete mitogenome sequences from PhyloTree and MitoTool (http://www.mitotool.org/),14 sequences of indigenous Americans were obtained from Mizuno et al.,2 Tamm et al.,15 Achilli et al.,16 Perego et al.,17 Kumar et al.18 We used the sequences that belong to haplogroups A, B, C and D. Sequence alignment was performed using MAFFT (http://mafft.cbrc.jp/alignment/software/).19 Imputation was carried out using a software by Huang et al.12 (http://www.ncgr.ac.cn/RiceHapMap). As there is no need to consider recombination in mitochondrial genome, we set a large window-size (w=500). For other parameters (p: penalty for different genotype in pairwise sequence similarity calculation, k: number of k-th highest similar sequences used for imputation and f: allele frequency threshold to determine genotype), we used the best set of parameters shown by Huang et al.12 (p=−7, k=5, f=0.7). To make sure, we evaluated imputation accuracy by using different k values (k=4, 5 and 7), and both k=4 and 5 showed the highest accuracy. Furthermore, we examined the reliability of imputation algorithm using simulated data. We produced missing data sets by randomly lacking 10, 20, 30, 40 and 50% of the nucleotides for each of randomly selected mitogenome sequences. Then, we conducted imputation and evaluated the sensitivity (filling rate) and specificity (accuracy). Filling rate was calculated as the percentage of nucleotides inferred and accuracy was defined as the percentage of nucleotides correctly inferred. We repeated this procedure 100 times.
Mitogenome sequence from 1500-year-old human remains
DNA was extracted from a human sacrifice (PPL99_3A) excavated at the Moon Pyramid in the Teotihuacan archeological site, Mexico.20 During all of the steps for DNA extraction, purification, and NGS library construction, we took all possible precautions to prevent contamination. The experiments were performed in a laboratory that is dedicated exclusively to ancient DNA work, which is physically isolated from other molecular research laboratories. All manipulations were performed in a laminar flow cabinet that is routinely irradiated with ultraviolet light. Frequent surface cleaning was performed routinely before and after working. A facemask, head cap and clean laboratory coat were always worn. Gloves were replaced frequently. All of the procedures were performed using gamma ray-irradiated disposable tubes and filter pipette tips. All non-disposable glasses and metallic materials were dry heat-sterilized at 160 °C for 2–6 h. All the reagents were molecular biology grade and ultrapure water was used. A series of NGS libraries was constructed by emulsion PCR and target enrichment according to our previously described protocol.6 The library was sequenced in 2 × 101 cycle runs on an Illumina HiSeq1500. Adapter sequences were trimmed using cutadapt21 and the reads measuring <28 nt in length were removed. The Burrows–Wheeler Aligner (BWA) does not consider the circularity of the mitogenome. Then, we joined the chrM sequence from the UCSC hg19 assembly to a haplogroup L sequence with a 9-bp deletion at nucleotide positions (np) 8281–8289 (accession number: EU092665) and we used this as a reference sequence for mapping. The sequence reads obtained were mapped to the reference mitogenome sequence using BWA with the default parameters.22 The mapped sequence reads were aligned with the hg19 sequence using Novoalign (Novocraft, http://novocraft.com/) with the default parameters. After alignment, duplicated reads were removed using Picard (http://picard.sourceforge.net). SNPs were identified using SAMtools.23
To examine the authenticity of the ancient DNA sequences obtained using NGS techniques, we considered two types of DNA degradation pattern, which differ between authentic ancient DNA and contamination with modern DNA. The first was the increased misincorporation frequency of thymine residues at positions where a cytosine is found in the reference sequence at the 5′-end of the mapped sequence reads, as well as the increased misincorporation frequency of adenine residues at positions where a guanine is found in the reference sequence at the 3′-end of the mapped sequence reads. The second was the increased frequency of purine residues at one base upstream of the 5′-end of the mapped sequence reads, as well as the increased frequency of pyrimidine residues at one base downstream of the 3′-end of the mapped sequence reads. The former is due to nucleotide misincorporation caused by the deamination of cytosine residues into uracil, a chemical analog of thymine, whereas the latter is due to depurination as a driving force of postmortem DNA fragmentation. The sequence reads obtained exhibited typical patterns of postmortem DNA degradation, which were consistent with previous studies.6, 24, 25, 26, 27, 28
Results
The use of a larger and more diverse reference panel is considered to improve the accuracy of imputation.29 However, the computational burden is a major concern, especially that incurred for the alignment step of imputation. To overcome this problem, we used nucleotide sequences at variant sites alone and not the entire mitogenome sequences. According to genome-wide association studies, the supplementation of missing data by imputation depends greatly on the panels used.30 We used different types of panel (mitogenome data file) to compare the results of imputation, that is, a panel comprising all of the 29 major haplogroups from around the world, a panel comprising worldwide mitogenome sequences belonging to the estimated haplogroup alone, a panel comprising mitogenome sequences from the population related most closely to an individual under investigation, and a panel comprising mitogenome sequences belonging to the estimated haplogroup from the population related most closely to an individual under investigation.
For 1500-year-old human remains (PPL99_3A) excavated at the Moon Pyramid in the Teotihuacan archeological site, Mexico, 89.3% of the mitogenome sequence was covered by 786 non-duplicated unique mapping reads with quality scores >20. On the basis of the available nucleotides, its haplotype was assumed to be A2, although both the diagnostic site of haplogroup A (np 663 of the revised Cambridge Reference Sequence) and three positions (np 16 290, 16 319 and 16 362) in the hypervariable segment 1 (HVS1) were missing. The average depth of coverage was 4.0-fold and 76.3% (12 640 sites) of the mitogenome sequence was covered by at least two non-duplicated reads with no discrepancies. However, 3931 nucleotides (sites) could not be determined for the following reasons: the depth of coverage was <2, the quality score was <20, or there was discordance in the sequence among the reads obtained, where they accounted for 23.7% of the whole mitogenome sequence (3931 sites) and we designated them as missing nucleotides. The haplogroup of mitogenome sequence of PPL99_3A was estimated after checking the validity of all sites with Phylotree Build 17.13
When we used Panel 1 (worldwide), 3927/3931 sites were filled in, thereby excluding four sites: np 152, 248, 10 873 and 15 301. Imputation using a worldwide haplogroup A panel (Panel 2) filled in 3930 sites but one site remained as not imputed. In addition, using a panel of indigenous American mitogenome sequences (Panel 3), 3930 sites were filled in a similar manner to Panel 2, thereby excluding one site, but their missing positions were different: np 153 and 152 for Panels 2 and 3, respectively. A panel comprising indigenous American haplogroup A mitogenome sequences (Panel 4) filled in all of the missing 3931 sites. As a result, the numbers of remaining missing sites were four, one, one and zero for Panels 1, 2, 3 and 4, respectively (Table 1). Thus, the number of missing sites can be reduced in an efficient manner by imputation. The missing sites, np663 of the haplogroup A diagnostic site and three positions (np 16 290, 16 319 and 16 362) in HVS1, were filled in with the expected nucleotides. We confirmed the imputation result by using PCR and direct sequencing, showing that np 663 was G (Supplementary Information).
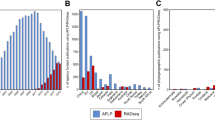
To validate the reliability of our imputation approach, we produced simulated data sets by randomly lacking 10, 20, 30, 40 and 50% of the nucleotides for each of the randomly selected mitogenome sequences for Panels 2 (worldwide haplogroup A mitogenome sequences), 3 (indigenous American mitogenome sequences) and 4 (indigenous American haplogroup A mitogenome sequences), respectively. As shown in Figure 1, the accuracy was highest for all the missing data when using Panel 4, although the filling rate using Panel 4 was lower than those using Panels 2 and 3. The filling rate using Panel 3 was relatively high, but the accuracy is lowest for all the missing data. Especially, the degree of deterioration in the accuracy was rapidly decreased, dependent on the missing percentages of the nucleotides. This might be due to its higher sequence diversity (due to the sequences consisting of haplogroups A, B, C and D), compared with those of Panels 2 and 4 (due to sequences consisting of haplogroup A alone).

Validation of the imputation approach obtained using simulated data. Filling rate (sensitivity: closed square) was calculated as the percentage of nucleotides inferred and accuracy (specificity: open circle) was defined as the percentage of nucleotides correctly inferred. 10%N, 20%N, 30%N, 40%N, and 50%N designate missing data that randomly lacks 10, 20, 30, 40, and 50% of the nucleotides, respectively. (a) Worldwide haplogroup A mitogenome sequences (Panel 2), (b) indigenous American mitogenome sequences (Panel 3) and (c) indigenous American haplogroup A mitogenome sequences (Panel 4). The values are the mean of 100 times of trials.
Discussion
The results of imputation were quite different according to the panels used (Table 2). We found that np 248, 10 873 and 15 301, which were still missing after imputation by Panel 1, were filled in using Panels 2, 3 and 4. In addition, there were no discrepancies in the nucleotides imputed. However, the nine sites filled in using Panel 1 (np 4248, 4824, 8794, 12 007, 16 111, 16 183, 16 290, 16 319 and 16 362) were filled in with different nucleotides when using Panels 2, 3 and 4, but the nucleotides were identical with these three panels. Intriguingly, Panels 2 and 4 successfully filled in np 152, whereas Panels 1 and 3 failed. For np 146, 235 and 663, the imputed nucleotides differed between Panels 1/3 and 2/4, that is, 146T, 235A and 663A by Panels 1/3, and 146C, 235G and 663G by Panels 2/4. The nucleotide at np 152 is known to vary among haplogroups. Furthermore, np 146 is a hotspot,31 while 235A and 663A are observed more commonly among haplogroups than 235G and 663G (663G is a diagnostic nucleotide for haplogroup A). The failure/success of imputation and the inconsistency of the nucleotides imputed according to the panels used are probably attributable to frequent parallel mutations. The use of a haplogroup-specific panel was highly advantageous for imputation. Imputation using Panels 1 and 3 filled in np 153 with nucleotide A, whereas that using Panel 4 added G; moreover, Panel 2 failed at imputation. The nucleotide at np 153 also varies among haplogroups, where 153A is observed more commonly among haplogroups than 153G. Indeed, 153G is one of the characteristic variant sites for sub-haplogroup A2,32 which probably explains why the worldwide haplogroup A panel failed at imputation. The missing np153 was imputed as nucleotide G using the indigenous American haplogroup A panel; therefore, PPL99_3A was assigned to sub-haplogroup A2. Haplogroup A of the indigenous American people is classified into sub-haplogroup A2, which is consistent with our result.
Together with the results of simulation analysis, our results showed that imputation using a common ancestral panel comprising mitogenome sequences belonging to the estimated haplogroup from the population related most closely to an individual under investigation provided more valid and reliable results than imputation using a panel that comprised all of the major haplogroups from around the world, a panel comprising worldwide mitogenome sequences belonging to the estimated haplogroup alone, and a panel comprising mitogenome sequences from the population related most closely to an individual under investigation. A larger and more diverse reference panel is thought to ensure more accurate imputation,29 but the present study demonstrated the risk of deriving genome sequence that can be imputed incorrectly due to recurrent mutations. The mitogenome contains highly mutable nucleotides called hotspots, which cause recurrent mutations.31 Due to the recurrent mutation of nucleotides, there is a possibility that missing nucleotides will be imputed incorrectly. However, by estimating the possible haplogroup at the initial step, it is possible to perform imputation more effectively. The use of an appropriate panel is essential. Thus, employing mitogenome sequences belonging to the estimated haplogroup from the population related most closely to an individual under investigation will obtain the best imputation results.
References
Smith, D. R. The past, present and future of mitochondrial genomics: have we sequenced enough mtDNAs? Brief Funct. Genomics 15, 47–54 (2016).
Mizuno, F., Gojobori, J., Wang, L., Onishi, K., Sugiyama, S., Granados, J. et al. Complete mitogenome analysis of indigenous populations in Mexico: its relevance for the origin of Mesoamericans. J. Hum. Genet. 59, 359–367 (2014).
Gojobori, J., Mizuno, F., Wang, L., Onishi, K., Granados, J., Gomez-Trejo, C. et al. mtDNA diversity of the Zapotec in Mexico suggests a population decline long before the first contact with Europeans. J. Hum. Genet. 160, 557–559 (2015).
Adler, C. J., Haak, W., Donlon, D. & Cooper, A. Survival and recovery of DNA from ancient teeth and bones. J. Arch. Sci. 38, 956–964 (2011).
Reed, F. A., Kontanis, E. J., Kennedy, K. A. & Aquadro, C. F. Brief communication: ancient DNA prospects from Sri Lankan highland dry caves support an emerging global pattern. Am. J. Phys. Anthropol. 121, 112–116 (2003).
Kihana, M., Mizuno, F., Sawafuji, R., Wang, L. & Ueda, S. Emulsion PCR-coupled target enrichment: an effective fishing method for high-throughput sequencing of poorly preserved ancient DNA. Gene 528, 347–351 (2013).
Hofreiter, M., Paijmans, J. L., Goodchild, H., Speller, C. F, Barlow, A., Fortes, G. G et al. The future of ancient DNA: technical advances and conceptual shifts. BioEssays 37, 284–293 (2015).
Bentley, D. R., Balasubramanian, S., Swerdlow, H. P., Smith, G. P., Milton, J., Brown, C. G. et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456, 53–59 (2008).
Marchini, J. & Howie, B. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 11, 499–511 (2011).
Pasaniuc, B., Rohland, N., McLaren, P. J., Garimella, K., Zaitlen, N., Li, H. et al. Extremely low-coverage sequencing and imputation increases power for genome-wide association studies. Nat. Genet. 44, 631–635 (2012).
Zheng, H. F., Rong, J. J., Liu, M., Han, F., Zhang, X. W., Richards, J. B. et al. Performance of genotype imputation for low frequency and rare variants from the 1000 genomes. PLoS ONE 10, e0116487 (2015).
Huang, X., Wei, X., Sang, T., Zhao, Q., Feng, Q., Zhao, Y. et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 42, 961–967 (2010).
van Oven, M. & Kayser, M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum. Mutat. 30, E386–E394 (2008).
Fan, L. & Yao, Y. G. MitoTool: a web server for the analysis and retrieval of human mitochondrial DNA sequence variations. Mitochondrion 11, 351–356 (2011).
Tamm, E., Kivisild, T., Reidla, M., Metspalu, M., Smith, D. G., Mulligan, C. J. et al. Beringian standstill and spread of Native American founders. PLoS ONE 2, e829 (2007).
Achilli, A., Perego, U. A., Bravi, C. M., Coble, M. D., Kong, Q. P., Woodward, S. R. et al. The phylogeny of the four pan-American MtDNA haplogroups: implications for evolutionary and disease studies. PLoS ONE 3, e1764 (2008).
Perego, U. A., Angerhofer, N., Pala, M., Olivieri, A., Lancioni, H., Hooshiar, K. B. et al. The initial peopling of the Americas: a growing number of founding mitochondrial genomes from Beringia. Genome Res. 20, 1174–1179 (2010).
Kumar, S., Bellis, C., Zlojutro, M., Melton, P. E., Blangero, J. & Curran, J. E. Large scale mitochondrial sequencing in Mexican Americans suggests a reappraisal of Native American origins. BMC Evol. Biol. 11, 293 (2011).
Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002).
Sugiyama, S. & Luján, L. Dedicatory burial/offering complexes at the moon pyramid, Teotihuacan. Ancient Mesoamerica 18, 127–146 (2007).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J 17, 10–12 (2011).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2009).
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Briggs, A. W., Stenzel, U., Johnson, P. L., Green, R. E., Kelso, J., Prüfer, K. et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl Acad. Sci. USA 104, 14616–14621 (2007).
Gilbert, M. T., Binladen, J., Miller, W., Wiuf, C., Willerslev, E., Poinar, H. et al. Recharacterization of ancient DNA miscoding lesions: insights in the era of sequencing-by-synthesis. Nucleic Acids Res. 35, 1–10 (2007).
Keller, A., Graefen, A., Ball, M., Matzas, M., Boisguerin, V., Maixner, F. et al. New insights into the Tyrolean Iceman’s origin and phenotype as inferred by whole-genome sequencing. Nat. Commun. 3, 698 (2012).
Krause, J., Fu, Q., Good, J. M., Viola, B., Shunkov, M. V., Derevianko, A. P. et al. The complete mitochondrial DNA genome of an unknown hominin from southern Siberia. Nature 464, 894–897 (2010).
Orlando, L., Ginolhac, A., Raghavan, M., Vilstrup, J., Rasmussen, M., Magnussen, K. et al. True single-molecule DNA sequencing of a Pleistocene horse bone. Genome Res. 21, 1705–1719 (2011).
Howie, B., Marchini, J. & Stephens, M. Genotype imputation with thousands of genomes. G3 (Bethesda) 1, 457–470 (2011).
Huang, G. H. & Tseng, Y. C. Genotype imputation accuracy with different reference panels in admixed populations. BMC Proc 8, S64 (2014).
Soares, P., Ermini, L., Thomson, N., Mormina, M., Rito, T., Röhl, A. et al. Correcting for purifying selection: an improved human mitochondrial molecular clock. Am. J. Hum. Genet. 84, 740–759 (2009).
Bandelt, H. J., Herrnstadt, C., Yao, Y. G., Kong, Q. P., Kivisild, T., Rengo, C. et al. Identification of Native American founder mtDNAs through the analysis of complete mtDNA sequences: some caveats. Ann. Hum. Genet. 67, 512–524 (2003).
Acknowledgements
This study was supported by JSPS KAKENHI Grant Number 25291104 (to SU), Qian Jiang Distinguished Professor program, State Key Development Program for Basic Research of China, 973 Program (No.2014CB541701), and Zhejiang Provincial Natural Science Foundation of China Grant (No. LZ13H02001; to LW). We thank Rikai Sawafuji and Koji Ishiya for processing of HiSeq1500 data.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Mizuno, F., Kumagai, M., Kurosaki, K. et al. Imputation approach for deducing a complete mitogenome sequence from low-depth-coverage next-generation sequencing data: application to ancient remains from the Moon Pyramid, Mexico. J Hum Genet 62, 631–635 (2017). https://doi.org/10.1038/jhg.2017.14
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2017.14
