
Abstract
Major advances in DNA sequencing technology have generated large quantities of sequence data that promote the development of statistical methods for rare variant association analyses. Although many population-based case control methods have been well developed for rare variant analysis, little work focuses on family-based studies. In this paper, we extend the existing methods to test for association of rare variants with case-parents data. We investigated the influence of non-variants and effects of causal variants on max- , multi-marker test, and collapsing method, and proposed an adaptive strategy based on a difference vector. Using simulations we show that the collapsing method is affected profoundly by the number of non-causal variants and different direction effects of causal variants and multi-marker test is most robust to non-causal variants and effects of causal variants. Our selective-difference strategy can improve power especially for collapsing method.
, multi-marker test, and collapsing method, and proposed an adaptive strategy based on a difference vector. Using simulations we show that the collapsing method is affected profoundly by the number of non-causal variants and different direction effects of causal variants and multi-marker test is most robust to non-causal variants and effects of causal variants. Our selective-difference strategy can improve power especially for collapsing method.
Similar content being viewed by others


Introduction
In the past few years, large genome-wide association (GWA) studies have uncovered a large number of common genetic variants involved in common diseases. However, most associations discovered in GWA studies only explained a limited proportion of heritability for most complex traits.1 Recently many resequencing based studies of candidate genes suggest many rare genetic variants contribute to the missing heretability unexplained by discovered common variants (CVs). Rare variants (RVs) are alternative forms of a gene that are present with a minor allele frequency (MAF) of <1%. Low frequencies of RVs make it difficult to detect RV association with approaches used for analysis of CVs.
The rapid advancement in DNA sequencing technology and the availability of large quantities of sequence data on large numbers of individuals provide an unprecedented opportunity to develop novel statistical methods for rare variant association analyses. Recently, the collapsing strategy has been widely adopted to analyse RVs. This strategy is to collapse all RVs across a causal region into a ‘super’ variant and then collectively test their association effect as a whole. Many statistical methods based on collapsing strategy have been recently developed. These include the cohort allelic sums test (CAST),2 the combined multivariate and collapsing method (CMC),3 the weighted-sum method4 and the variable threshold method.5 These methods, with the assumption that all variants in a region have an effect on the phenotype and the effects are in the same direction with the same magnitude, can improve power by combining information of multiple RVs. However, these tests will lose power when the set of collapsed variants includes non-causal variants or the effects of causal variants have different directions. Various methods have been proposed recently to overcome these limitations. These include C-alpha score test,6 the sequence kernel association test,7 and the adaptive sum strategy.8 The series of adaptive tests proposed by Pan and Shen8 can be considered as the extension of the variable threshold method. The former is based on the frequency of the minor allele, while the latter is to order the standardised magnitudes of a statistic U or the locations of their corresponding RVs.
Although many methods have been well developed for rare variant analysis, relatively little work has focused on family-based studies. Compared with population-based case control studies, family-based studies are more attractive due to their robustness to population stratification which is more prominent for rare variants.9 Moreover, because of using information about transmission of genetic factors within families, family-based methods for single SNP association are potentially more powerful than the population-based methods for rare diseases.10, 11 In family-based analysis, one way is to transform the family-based data and apply case–control statistical tests. The commonly used strategy is to use nontransmitted genotypes as control (also named as pseudocontrols or complements) of affected offspring in case-parents data and construct a difference vector calculated by comparing the genotypes of affected offspring with their corresponding ‘complements’.12 In this paper, we will extend the existing methods including max- , multi-marker test, and collapsing method to test for association of rare variants with family-based study and, based on the difference vector, use an adaptive strategy to eliminate the influence of non-causal variants and effects of causal variants. In our method, we choose RVs according to the magnitude of difference. Through simulation studies, we will assess the type I error rates and the power.
, multi-marker test, and collapsing method to test for association of rare variants with family-based study and, based on the difference vector, use an adaptive strategy to eliminate the influence of non-causal variants and effects of causal variants. In our method, we choose RVs according to the magnitude of difference. Through simulation studies, we will assess the type I error rates and the power.
Materials and methods
We consider a sample of n trios-two parents and an affected offspring in each family. The variants and the triads are indexed by i(i=1, 2, ⋯, k) and j (j=1, 2, ⋯, n), respectively. Let Mij, Fij and Oij be the number of copies of minor alleles carried by the mother, father and offspring, respectively, in the jth trio at the ith variant. Let δij=2Oij−Fij−Mij. δij presents the difference in genotypes between the affected offspring and the complement for the jth trio at the ith variant. Here, missing individual variant genotype is permitted, that means, the genotypes at some variants can be sporadically unknown for a member in the family. We define a family as a informative family for variant i when the genotypes are known for each member of the trio and δij≠0. Let ni(ni≤n) be the number of the informative families and  be the sample mean of δij among informative families at the ith variant. Denote the variance of
be the sample mean of δij among informative families at the ith variant. Denote the variance of  by
by  , where
, where  .
.
Family-based association test
A simple approach for family-based association test (FBAT) is to analyse individual variants separately. For the ith variant, define

Under the null hypothesis of no association Zi is approximately N(0,1). A Bonferroni correction is used for k tests when all rare variants are independent. If we take into account the correlation between the variants, the maximum of the  across all k variants can be adopted with a permutation procedure.12 The permutation procedure is as follows. We first calculate the data-based statistic. Then we recalculate permutation-based statistic by random reassigning the labels ‘case’ and ‘complement’ with equal probability. We repeat this process B times and then the P-value is estimated as the proportion of permutation-based statistics that are larger than the data-based statistic.
across all k variants can be adopted with a permutation procedure.12 The permutation procedure is as follows. We first calculate the data-based statistic. Then we recalculate permutation-based statistic by random reassigning the labels ‘case’ and ‘complement’ with equal probability. We repeat this process B times and then the P-value is estimated as the proportion of permutation-based statistics that are larger than the data-based statistic.
Another approach for the FBAT is Multi-Marker test, which is to test all variants simultaneously with the use of a multivariate test.3 However, it requires the genotypes known for each member of the triads at k variants. Assume that the genotypes of n case-parents triads are available. Define a k-dimensional random vector δ=(δ1, δ2,⋯, δk)T be the difference vector for k variants. Then  is the sample mean vector and
is the sample mean vector and  is the covariance matrix of
is the covariance matrix of  when rare variants are independent. The multi-marker test is then given by
when rare variants are independent. The multi-marker test is then given by

In large samples, TM has an asymptotically χ2 distribution with degrees of freedom equal to the rank of Σ.
Collapsing method for rare variants
Collapsing method is to ‘collapse’ multiple variants into a single variant in a gene or region. We assume that all variants have an effect with the same direction on disease susceptibility. Let  , then
, then  . The standardised test statistic is
. The standardised test statistic is

Under the null hypothesis of no association ZC is approximately N(0,1).
The original collapsing approach collapses all the variants in the region of interest and does not eliminate the noise generated by the non-causal variants. In order to remove the influence of the non-causal variants, we propose to use an adaptive strategy based on the difference of  . We sort k variants in ascending order of
. We sort k variants in ascending order of  and let
and let  be a set containing all k ordered variants. Let
be a set containing all k ordered variants. Let 
 be the set which delete the first s variants from G, for example, G(0)=G,
be the set which delete the first s variants from G, for example, G(0)=G,  , and
, and  . We obtain k variant sets G(0), G(1),⋯, G(k−1) containing k, k−1,⋯,1 variants, respectively. The values of
. We obtain k variant sets G(0), G(1),⋯, G(k−1) containing k, k−1,⋯,1 variants, respectively. The values of  in G(s) are larger than those in variant sets ahead of G(s). For each G(s), we calculate the statistic, denoted by ZG(s), with collapsing method,
in G(s) are larger than those in variant sets ahead of G(s). For each G(s), we calculate the statistic, denoted by ZG(s), with collapsing method,

Our test statistic, here, denoted as max-ZG, is the maximum of the ZG(s), that is, max- . The variant set corresponding to the max-ZG can be considered as the optimal set containing variants associated with disease. We also denote the statistic corresponding to TM based on this selective-difference strategy as max-TM,
. The variant set corresponding to the max-ZG can be considered as the optimal set containing variants associated with disease. We also denote the statistic corresponding to TM based on this selective-difference strategy as max-TM,

where, TMG(s) corresponds to the statistic TM calculated with equation (2) in variant set G(s). The statistical significance can be assessed by permutation.
Results
Simulation setting
To assess the performance of these statistics, we perform the simulation study under a wide range of parameter values (the program is available on request). The simulation parameter includes the number of variants, the MAF at each variant, the number and effect size of causal variants, and the sample size. We consider k (k=10, 20, 50) variants in the region and the proportion of non-causal variants are 20%, 40%, 60% and 80% (here, let q be the number of causal variants). We assume that variants are independent and firstly create parental haplotypes and then generate offspring haplotypes. Remember that although haplotypes are simulated in our study, only genotype data are used. The disease status for an individual’s phenotype is determined by the following logistic model:13

where c is a background chance of being affected for a subject with no minor alleles, ORi is the effect size of variant i and Oij is the number of copies of minor alleles at the ith variant. The parameters are chosen as follows: c=0.01. The minor allele frequencies of all variants are randomly determined with values ranging from 0.001 to 0.01. OR=1 for all variants under the null hypothesis of no association. Under the alternative hypothesis of association, we consider three scenarios: scenario A is that variants associated with disease have the same OR value, scenario B is that variants associated with disease have the same positive direction but different effects, and scenario C is that variants associated with disease have different direction effects. In scenario A, we let OR=2 for causal variants. In scenario B, we let OR∈[1.2, 3] with increments of  for causal variant 1 to variant q. In scenario C, we let OR∈[1.2, 3] for half of causal variants and OR∈[0.2, 0.8] for the rest causal variants. In three scenarios, OR=1 for non-causal variants. We assume that the genotypes of each individual for all variants are available in the analysis. The number of case-parent triads, n, is chosen as 500, 1000 and 1500.
for causal variant 1 to variant q. In scenario C, we let OR∈[1.2, 3] for half of causal variants and OR∈[0.2, 0.8] for the rest causal variants. In three scenarios, OR=1 for non-causal variants. We assume that the genotypes of each individual for all variants are available in the analysis. The number of case-parent triads, n, is chosen as 500, 1000 and 1500.
In each simulation scenario, we calculate the values of the statistics according to whether we use or not use selective-difference strategy. When not using selective-difference strategy, we consider the statistics  , the multi-marker test TM, and the statistic ZC with collapsing method. When using selective-difference strategy, we consider the multi-marker test max-TM and the collapsing statistic max-ZG. P-values of these statistics are estimated as the proportion of the permutation-based statistics that are larger than the data-based statistic by 5000 (B=5000) permutations. Type I error rates and powers are the proportion of p-values that are less than a significance level of 0.05 in 1000 replications when the null hypothesis/the alternative hypothesis holds.
, the multi-marker test TM, and the statistic ZC with collapsing method. When using selective-difference strategy, we consider the multi-marker test max-TM and the collapsing statistic max-ZG. P-values of these statistics are estimated as the proportion of the permutation-based statistics that are larger than the data-based statistic by 5000 (B=5000) permutations. Type I error rates and powers are the proportion of p-values that are less than a significance level of 0.05 in 1000 replications when the null hypothesis/the alternative hypothesis holds.
Type I error rate and power
We present in Table 1 the estimated type I error rates for sample sizes from 500 to 1500 individuals. As shown in Table 1, the type I error rates are all around the nominal levels.
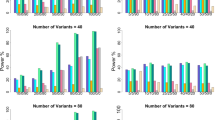
The power estimates are exhibited in Tables 2, 3, 4 for three scenarios, respectively, when the sample size is 500. From Tables 2,3, it is found that the power estimates of all tests decrease with the increasing of the number of non-causal variants for a given number of variants, indicating that the powers of these tests are affected by non-causal variants. We can see that the multi-marker test is least affected and the collapsing method is most affected by non-causal variants. For example, when there are 10 variants in scenario A, with the number of non-causal variants increasing from 6 to 8, the powers of TM and ZC decrease from 0.965% to 0.866% and 0.632% to 0.200%, with 10.26% and 68.35% decline rate, respectively. Nevertheless, this difference becomes less severe by adopting selective-difference strategy. It can be seen that the multi-marker test has highest power and powers of max-TM with selective-difference strategy are slightly larger than those of TM. We observed that powers of collapsing method with selective-difference strategy are larger than those not with selective-difference strategies, especially for the large number of non-causal variants. When the number of variants is 10, powers of collapsing method with selective-difference strategy are very close to those of multi-marker test.
It can be seen from Table 4 that, when causal variants have different direction effects, the collapsing method has very low power. However, powers of collapsing method are improved by using selective-difference strategy, and especially when the number of variants is 10, powers of collapsing method are sharply improved from ~10% to >90%. We also observed that, similar to those under the first two scenarios, powers of multi-marker test are largest and can be improved by selective-difference strategy. The results in Table 4 showed that the collapsing method has been affected profoundly by different direction effects of causal variants and selective-difference strategy can largely enhance the power, and at the same time, multi-marker test is most robust to different direction effects of causal variants. Furthermore, we can see that powers for all statistic tests decease with the number of variants increasing.
We also investigated the performance of our method in the presence of population stratification. We assume that the study population is composed of two subpopulations both with 50%. In the two subpopulations, the minor allele frequencies of all variants are uniformly generated between 0.001 and 0.01. OR=1 for all variants under the null hypothesis. In the first subpopulation, c=0.01, the values of OR vary from 1.2 to 3.0 with increments of  for causal variant 1 to variant q and OR=1 for non-causal variants under the alternative hypothesis of association. In the second subpopulation, c=0.008, the values of OR vary from 1.2 to 2.0 with increments of
for causal variant 1 to variant q and OR=1 for non-causal variants under the alternative hypothesis of association. In the second subpopulation, c=0.008, the values of OR vary from 1.2 to 2.0 with increments of  for causal variant 1 to variant q and OR=1 for non-causal variants under the alternative hypothesis of association. we found that type I error rates are well controlled (data not shown). The results for the power are similar to those under the homogeneous population (data not shown). In addition, we explored the effects of different sample sizes on the power of these statistics. As expected, the power increases when the sample size is increased (data not shown).
for causal variant 1 to variant q and OR=1 for non-causal variants under the alternative hypothesis of association. we found that type I error rates are well controlled (data not shown). The results for the power are similar to those under the homogeneous population (data not shown). In addition, we explored the effects of different sample sizes on the power of these statistics. As expected, the power increases when the sample size is increased (data not shown).
Computation time
The computation time for these statistics using the selective-difference strategy depends on the number of variants, the sample size and the permutation time. To analyse 10 variants on 500, 1000 and 1500 case-parents trios with 5000 permutations requires 0.5, 1.2 and 2 min, respectively. Analysing 20 variants on 500, 1000 and 1500 case-parents trios with 5000 permutations requires 1, 1.8 and 2.5 min, respectively. In addtion, 1.6, 2.3 and 3 min are required for 50 variants on 500, 1000 and 1500 case-parents trios with 5000 permutations, respectively.
Discussion
In this paper, we extended the existing methods including  , multi-marker test, and collapsing method to test RVs association with disease susceptibility using case-parents data. We used case-parents triad to create the genotype difference between affected offspring with their corresponding ‘complements’ and adopted a selective-difference strategy by ordering the means of the differences for all variants. Our method can be considered the extension of the adaptive methods proposed by Price et al.5 and Pan and Shen.8 However, at least two characteristics of our method are totally different from their methods: (1) our method uses the case-parents data and offers a substantial benefit of being robust to admixture population, while their methods are for case control population-based analysis, (2) our approach is based on the order of the means of the differences between affected offspring with their corresponding ‘complements’, whereas the method of Price5 uses the frequency of the minor allele and the method of Pan and Shen8 is to order the standardised magnitudes of a statistic U or the locations of their corresponding RVs. We assessed the performance of our method by simulation analysis.
, multi-marker test, and collapsing method to test RVs association with disease susceptibility using case-parents data. We used case-parents triad to create the genotype difference between affected offspring with their corresponding ‘complements’ and adopted a selective-difference strategy by ordering the means of the differences for all variants. Our method can be considered the extension of the adaptive methods proposed by Price et al.5 and Pan and Shen.8 However, at least two characteristics of our method are totally different from their methods: (1) our method uses the case-parents data and offers a substantial benefit of being robust to admixture population, while their methods are for case control population-based analysis, (2) our approach is based on the order of the means of the differences between affected offspring with their corresponding ‘complements’, whereas the method of Price5 uses the frequency of the minor allele and the method of Pan and Shen8 is to order the standardised magnitudes of a statistic U or the locations of their corresponding RVs. We assessed the performance of our method by simulation analysis.
In our simulations, we investigated the influence of non-causal variants and the effect size of causal variants on the power. The results showed that powers of these methods are all affected by the number of non-causal variants and the effect size of causal variants. Here, we found that the collapsing method is affected profoundly by the number of non-causal variants and different direction effects of causal variants and multi-marker test is most robust to non-causal variants and effects of causal variants. The selective-difference strategy can improve power especially for collapsing method. It should be noted that, although our method is designed for case-parents data, it is flexible in application. In practice, when multiple markers are studied, individuals may have incomplete information of individual marker data. Our strategy for collapsing method is capable of handling missing SNP data. We can also use single-parent families to obtain the difference and then construct these statistic tests for RVs analyses when we make a study of diseases of late onset.
References
Maher, B. Personal genomes: the case of the missing heritability. Nature 456, 18–21 (2008).
Morgenthaler, S. & Thilly, W. G. A strategy to discover genes that carry multiallelic or mono-allelic risk for common diseases: a cohort allelic sums test (CAST). Mutation Res. 615, 28–56 (2007).
Li, B. & Leal, S. M. Methods for detecting association with rare variants for common diseases: application to analysis of sequence data. Am. J. Hum. Genet. 83, 311–321 (2008).
Madsen, B. E. & Browning, S. R. A groupwise association test for rare mutations using a weighter sum statistic. PLoS Genet. 5, e1000384 (2009).
Price, A. L., Kryukov, G. V., Bakker, P. I. W., Purcell, S. M., Staples, J., Wei, L. J. et al. Pooled association tests for rare variants in exon-resequencing studies. Am. J. Hum. Genet. 86, 832–838 (2010).
Neale, B. M., Rivas, M. A., Voight, B. F., Altshuler, D., Devlin, B., Orho-Melander, M. et al. Testing for an unusual distribution of rare variants. PLoS Genet. 7, e1001322 (2011).
Wu, M. C., Lee, S., Cai, T., Li, Y., Boehnke, M. & Lin, X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 89, 82–93 (2011).
Pan, W. & Shen, X. T. Adaptive tests for association analysis of rare variants. Genetic Epidemiol. 35, 381–388 (2011).
Laird, N. M. & Lange, C. Family-based designs in the age of large-scale gene association studies. Nat. Rev. Genet. 7, 385–394 (2006).
Bansal, V., Libiger, O., Torkamani, A. & Schork, N. J. Statistical analysis strategies for association studies involving rare variants. Nat. Rev. Genet. 11, 773–785 (2010).
Gravel, S., Henn, B. M., Gutenkunst, R. N., Indap, A. R., Marth, G. T., Clark, A. G. et al. Demographic history and rare allele sharing among human populations. Proc. Natl Acad. Sci. USA 108, 11983–11988 (2011).
Shi, M., Umbach, D. M. & Weinberg, C. R. Identification of risk-related haplotypes with the use of multiple SNPs from nuclear families. Am. J. Hum. Genet. 81, 53–66 (2007).
Preston, M. D. & Dudbridge, F. Utilising family-based designs for detecting rare variant disease associations. Ann. Hum. Genet. 78, 129–140 (2014).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (11301206), Foundation of Hunan Educational Committee (16A166) and China Scholarship Council.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Li, YM., Xiang, Y. Detecting disease association with rare variants in case-parents studies. J Hum Genet 62, 549–552 (2017). https://doi.org/10.1038/jhg.2017.1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2017.1
