
Abstract
SLC1A4 deficiency is a recently described neurodevelopmental disorder associated with microcephaly, global developmental delay, abnormal myelination, thin corpus callosum and seizures. It has been mainly reported in the Ashkenazi–Jewish population with affected individuals homozygous for the p.Glu256Lys variant. Exome sequencing performed in an Irish proband identified a novel homozygous nonsense SLC1A4 variant [p.Trp453*], confirming a second case of SLC1A4-associated infantile spasms. As this is the first European identified, population ancestry analysis of the Exome Aggregation Consortium database was performed to determine the wider ethnic background of SLC1A4 deficiency carriers. p.Glu256Lys was found in Hispanic and South Asian populations. Other potential disease-causing variants were also identified. Investigation for SLC1A4 deficiency should be performed regardless of ethnicity and extend to include unexplained early-onset epileptic encephalopathy.
Similar content being viewed by others

Main
Three recent studies have identified a total of 15 patients with autosomal recessive disease-causing variants in the neutral amino acid (ASCT1/Na+-dependent transporter) gene SLC1A4 causing a severe early-onset neurodevelopmental disorder affecting brain serine metabolism.1, 2, 3 SLC1A4 deficiency is characterized by congenital/progressive microcephaly, severe/profound global developmental delay, neuroimaging abnormalities (abnormal myelination, thin corpus callosum) and seizures (Supplementary Table 1).1, 2, 3 Almost all affected individuals reported to date are homozygous for the p.Glu256Lys variant and of Ashkenazi–Jewish (AJ) descent (n=12) except for two AJ-Iraqi Jewish children (compound heterozygous)2, 3 and one Palestinian.2 To date, no other ethnicities affected by this disorder have been described. We report the first European (Irish, non-AJ) individual with a novel SLC1A4 disease-causing variant and review population ancestry information for known and predicted disease-causing variants.
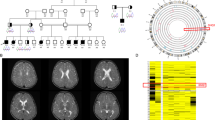
The male proband was born at term to consanguineous (third cousins) parents (weight=2.8 kg). On a background of neurodevelopmental delay and congenital microcephaly (0.4th centile), he presented at 5 months with an early-onset seizure disorder characterized by infantile spasms (electroencephalogram presented in Figure 1a), focal motor and focal dyscognitive seizures. Following failed treatments with several antiepileptic drugs, seizures ceased eventually on a combination of clobazam and vigabatrin (18 months). Severe progressive microcephaly (0.4th centile (32 cm) at birth to below the 0.4th centile) with hypotonia and developmental delay were noted at presentation. At the age of 3 years, he remains hypotonic with brisk deep tendon reflexes and extensor plantar responses. Developmental progress has been minimal (not sitting unsupported or reaching, not babbling, no visual fixation). Brain magnetic resonance imaging shows a thin corpus callosum, progressive white matter volume loss and markedly delayed myelination (Figure 1b–d). Chromosomal microarray, MECP2 duplication and investigations for inborn errors of metabolism were unremarkable (further clinical details in Supplementary Information).

(a) Electroencephalogram (EEG) at presentation (5 months) showing modified hypsarrhythmia, that is, slow disorganized background, high-amplitude delta and frequent epileptiform spike and polyspike wave discharges (sensitivity reduced to 30 uV mm−1). (b) Magentic resonance imaging (MRI) brain axial T2 at 5 months showing delayed myelination and mild increase in ventricular fluid space. (c) MRI brain axial T2 at 2.25 years showing hypomyelination, white matter volume loss and further increase in ventricular size. (d) Saggital T1 MRI brain showing thin corpus callosum. A full color version of this figure is available at the Journal of Human Genetics journal online.
Informed parental consent was obtained to collect clinical data and perform whole-exome sequencing (performed at WuXiNextCODE; https://www.nextcode.com; Reykjavik, Iceland). A recessive inheritance analysis pipeline was adopted due to the consanguineous nature of the pedigree. A minor allele frequency (MAF) <0.5% and <0.0025% was chosen for homozygous and heterozygous variants, respectively. A genotype frequency of 0.0025% was selected for genes with ⩾2 variants, that is, possible compound heterozygous genes. Sanger sequencing was used for variant validation.
Filtering for rare and novel variants identified five homozygous/hemizygous variants (see Supplementary Table 3 for more information). Four of these variants were present on the X chromosome (SPANX1, MAGEA3, UPRT and MECP2). SPANX1 was eliminated as a candidate gene based on known gene function (spermiogenesis). The variants in MAGEA3 and URPT were also excluded as candidate disease-causing variants, as they are present in unaffected controls in the Exome Aggregation Consortium (ExAC) (143 and 48 individuals, respectively) and Exome Variant Server (EVS) (4 and 0 individuals, respectively).4, 5 One rare variant in MECP2 (rs61749739) has been previously reported as ‘benign’ in ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/variation/143659/).
The only recessive variant was a novel homozygous nonsense variant in SLC1A4 (chr2: 65245807, c.1358G>A) leading to a premature stop codon at position 453 (Trp453*, transcript ID: ENST00000234256). In the homozygous state, there is a complete absence of the final 80 amino acids (15%), leading to an assumed loss-of-function effect. This variant was absent in the ExAC database, the EVS database and our own private Irish control database (n=97).4, 5 Both parents were heterozygous for the variant (Supplementary Figure 1).
Due to the finding of SLC1A4 deficiency in the Irish (non-AJ) population, ancestry analysis using the ExAC database (n=67 006 individuals) was performed to review the ethnic background and types of SLC1A4 variants.4 The ExAC control population was examined for the presence of moderate-/high-impact SLC1A4 variants and data was assessed for global and ethnic MAF (Figure 2 and Supplementary Information). Five variant effect prediction programs were then used to assess the potential for each variant to cause damage to protein function or structure. Variants that were predicted to be damaging by all programs were designated ‘potentially’ disease causing.

Flow diagram showing approach to identification of known and predicted SLC1A4 disease-causing variants across ethnic groups in Exome Aggregation Consortium database (ExAC) (n=67,006 individuals). A full color version of this figure is available at the Journal of Human Genetics journal online.
A total of 151 medium-/high-impact SLC1A4 variants were identified in ExAC (Supplementary Table 2). Two published variants p.Glu256Lys (AJ) and p.Arg457Trp (Palestinian),2 both disease causing in the homozygous states, were present in ExAC in the heterozygous state in 21 and 19 individuals, respectively, across European (non-Finnish), Hispanic and Central South Asian ethnicities (Supplementary Figure 2). p.Arg457Trp was also found in other ethnic groups not specified. Of 151 medium-/high-impact variants, 49 were predicted to be damaging by all prediction tools. These include both p.Glu256Lys and p.Arg457Trp, and variants in the third sodium ion-binding site previously shown in experimental models to have an important role in SLC1A4 (ASCT1) structure and function.6 The only other published heterozygous disease-causing variant p.Leu315fs (Iraqi-AJ) and the novel Irish variant p.Trp453* were absent in ExAC.
This is the second individual with SLC1A4 deficiency with infantile spasms and the first European/Irish Caucasian. To date, SLC1A4 deficiency has been described in patients predominantly of AJ descent, with the p.Glu256Lys founder disease-causing variant in that population having a carrier frequency of 1/144.1, 2, 3 Although these reports have emphasized SLC1A4 deficiency as being particularly important in the AJ population,1, 2, 3 review of ExAC also demonstrates that 10% of individuals carrying the p.Glu256Lys allele in this control population are of Hispanic and South Asian descent. Furthermore, the homozygous p.Arg457Trp disease-causing variant found in one Palestinian proband so far was present with a similar MAF (0.0157%) to p.Glu256Lys (MAF=0.0165%), a potentially important disease variant.2 The majority of individuals (17/19) identified in ExAC carrying the p.Arg457Trp variant were Hispanic. MAFs of p.Arg457Trp in Hispanic and other ethnic groups in ExAC were 0.15% and 0.22%, respectively. Other potential disease-causing variants were also identified (Figure 2 and Supplementary Material).
The three patients so far reported with high-impact variants (patient II:3 [p. Leu315fs] of Heimer et al.3), patient 10 [p.Leu315fs] of Damseh et al.2 and this Irish patient [p.Trp453*], all present with a more severely affected developmental and seizure phenotype. This may be a consequence of the higher variant impact on protein function in these patients compared with Glu256Lys.
SLC1A4 deficiency should not be considered a population-specific disorder, and all patients with unexplained severe neurodevelopmental delay and the features outlined should be investigated regardless of ethnicity, as there are no known metabolic markers of this potentially treatable condition. This report, describing infantile spasms with SLC1A4 deficiency highlights the additional importance of this gene when investigating infants with unexplained early-onset epileptic encephalopathy.
References
Srour, M., Mandan, F. F., Gan-Or, Z., Labuda, D., Nassif, C., Oskoui, M. et al. A homozygous mutation in SLC1A4 in siblings with severe intellectual disability and microcephaly. Clin. Genet. 88, e1–e4 (2015).
Damseh, N., Simonin, A., Jalas, C., Picoraro, J. A., Shaag, A., Cho, M.T. et al. Mutations in SLC1A4, encoding the brain serine transporter, are associated with developmental delay, microcephaly and hypomyelination. J. Med. Genet. 52, 541–547 (2015).
Heimer, G., Marek-Yagel, D., Eyal, E., Barel, O., Oz Levi, D., Hoffmann, C. et al. SLC1A4 mutations cause a novel disorder of intellectual disability, progressive microcephaly, spasticity and thin corpus callosum. Clin. Genet. 88, 327–335 (2015).
Exome Aggregation Consortium,Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E. et al. Analysis of protein-coding genetic variation in 60,706 humans. bioRxiv. (e-pub ahead of print 30 October 2015; doi:10.1101/030338).
NHLBI GO Exome Sequencing Project Exome Variant Server (2016) http://evs.gs.washington.edu/EVS/. Accessed 7 October 2015.
Scopeelliti, A. J., Heinzelmann, G., Kuyucak, S., Ryan, R. M. & Vandenberg, R. J. Na+ interactions with the neutral amino acid transporter ASCT1. J. Biol. Chem. 289, 17468–17479 (2014).
Acknowledgements
We thank the parents for participation and consent for the analysis. We also thank the Children’s Fund for Health Ltd, Temple Street Children’s University Hospital, Dublin, Ireland for funding this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Conroy, J., Allen, N., Gorman, K. et al. Novel European SLC1A4 variant: infantile spasms and population ancestry analysis. J Hum Genet 61, 761–764 (2016). https://doi.org/10.1038/jhg.2016.44
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2016.44
This article is cited by
-
Genetic etiology of progressive pediatric neurological disorders
Pediatric Research (2024)
-
Generation and characterization of a knock-in mouse model for spastic tetraplegia, thin corpus callosum, and progressive microcephaly (SPATCCM)
Mammalian Genome (2023)
-
West syndrome: a comprehensive review
Neurological Sciences (2020)
-
Amino Acid Transporters and Exchangers from the SLC1A Family: Structure, Mechanism and Roles in Physiology and Cancer
Neurochemical Research (2020)

{kind=link}
{kind=link}