
Abstract
By using next-generation sequencing targeted to MitoExome including the entire mtDNA and exons of 1033 genes encoding the mitochondrial proteome, we described here a novel m.11240C>T mutation in the mitochondrial ND4 gene from a patient with Leigh syndrome. High mutant loads of m.11240C>T were detected in blood, urinary epithelium, oral mucosal epithelium cells, and skin fibroblasts of the patient. Decreased mitochondrial complex I activity was found in transmitochondrial cybrids containing the m.11240C>T mutation with biochemical analysis. Furthermore, functional investigations confirmed that mitochondria with the m.11240C>T variant exhibited lower adenosine triphosphate-related mitochondrial respiration. However, complex I assembly in mutant cybrids was not affected. While this mutation was located in the fourth hydrophobic trans-membrane region of ND4 gene, we suggested that mutation of m.11240C>T might impair the proton pumping channel of complex I but had little effect on the complex I assembly. In conclusion, we identified m.11240C>T as a novel mitochondrial disease-related mtDNA mutation.
Similar content being viewed by others

Introduction
Isolated respiratory chain complex I (CI, NADH:quinone oxidoreductase) deficiency was frequently (about 30% of cases) observed in patients with mitochondrial disorders.1 Pathological conditions such as exercise intolerance and developmental delay,2 leukoencephalopathy,3 Leber hereditary optic neuropathy,4 mitochondrial myopathy, lactic acidosis and stroke-like episodes,5 and cardiomyopathy were associated with CI deficiency.6 Mammalian mitochondrial CI is composed of 38 nuclear DNA encoded and 7 mtDNA encoded subunits. Mutations in seven mitochondrial DNA (mtDNA) encoded subunits, including ND1, ND2, ND3, ND4, ND5, ND6 and ND4L, were reported in patients with mitochondrial disorders.7
Leigh syndrome (LS), also referred to as sub-acute necrotizing encephalopathy, is a rare inherited neurological disease with common features of developmental delay, ataxia, progressive loss of mental/movement abilities and visible symmetrical lesions in basal ganglia via magnetic resonance imaging. Mutations in the mitochondrial DNA-encoded subunits of respiratory chain CI and its assembly factors were frequently reported in LS.8
By using next-generation sequencing targeted to MitoExome including the entire mtDNA and exons of 1033 genes encoding the mitochondrial proteome, new pathogenic mtDNA mutations were being frequently identified.9 However, the functional consequences in most of these mutations are currently not known. While this strategy is by far the most powerful tool to provide a molecular diagnosis of mitochondrial disorders, we performed mutation screening with previously established MitoExome sequencing technology in patients with LS. In one patient, we identified a novel heteroplasmic mutation of m.11240C>T [p.Leu161Phe] in NADH dehydrogenase 4 (ND4) gene. The potential pathogenicity of this CI mutation was assessed by determining the mitochondrial respiration, CI assembly and respiratory chain complexes activity in cybrids with and without m.11240C>T mutation.
Materials and methods
MitoExome sequencing
Genes encoding the entire predicted mitochondrial proteome (exon regions of 1033 mitochondria nuclear genes and the 16 569 bp mitochondria genome) were captured and sequenced on an Illumina HiSeq 2000 sequencer (Illumina, SanDiego, CA, USA) as described previously.10 The average reads and coverage of each targeted nuclear base and mtDNA base were described (Table 1). For sequencing analysis, Illumina clean reads were aligned to each human reference genome (GRCh38, UCSC) using the BWA program and quality scores were recalibrated and realigned to reference using the GATK software package. The pathogenicity of the variants were evaluated through the filters such as allele frequency, effects on protein function and consistent with a recessive model of pathogenesis.10
Generation of cell lines and culture conditions
About 2 ml venous blood was collected from the patient to obtain platelets. Trans-mitochondrial cybrids were obtained by fusion of mtDNA-less ρ0 human osteosarcoma 143B cells with platelets derived from the patient and his mother, as described previously.11 The transformant cybrids were cultured in high-glucose Dulbecco’s modified Eagle’s medium (Hyclone, Waltham, MA, USA) containing 10% cosmic calf serum (Gibco, Carlsbad, CA, USA).
mtDNA analysis
To test the mutant loads of mtDNA mutation, 1 ml venous blood and 15 ml urinary epithelium cells-containing urine from the patient and his maternal family members (mother, aunt and grandmother) were collected. Oral mucosal epithelium cells from the patient and his mother were collected using disposable oral cells collection swab. Also, a 0.2 × 0.2 cm skin biopsy from medial side of upper arm of the patient was obtained through outpatient surgery. Genomic DNA was extracted using an sodium dodecyl sulfate lysis protocol as described previously.12 Mutant loads of m.11240C>T were assessed by polymerase chain reaction (PCR)-restriction fragment length polymorphism (PCR-RFLP) or allele-specific amplification-based real-time PCR method. For real-time PCR method: the following allele-specific primers were used: mtDNA 11240C: forward, 5′-TAGTAGGCTCCCTTCCCCTAC-3′; mtDNA 11240T: forward, 5′-TAGTAGGCTCCCTTCCCCTAT-3′; reverse, 5′-GTGGAGTCCGTAAAGAGGTATC-3′. The PCR amplification efficiencies of these primers were between 90% and 110%. The mutant load of 11240T was calculated using the following equation: Percentage of 11240T=2(Ct11240C -Ct11240T)/(1+ 2(Ct11240C -Ct11240T)) × 100%.
For PCR-RFLP: the sequences of the primers used in this study were as follows: forward, 5′-AACGCCTGAACGCAGGCACATACTTCCTATTCTACACCCTAGTAGGCTCCCTTCCCGTA-3′; reverse, 5′-AGGGAGTCATAAGTGGAGTCCGTAAAGAGG-3′. A 230 bp segment of PCR product was subjected to complete digestion with Rsa I (NEB, New England Biolabs, Ipswich, MA,USA) at 37 °C. The wild type ND4 fragment was cut into two small fragments of 173 bp and 57 bp, while the mutant ND4 fragment remained intact. The intensity of the bands was quantified by Gel-Pro Analyzer 4.0 (MediaCybernetics, Warrendale, PA, USA).
Mitochondrial protein preparation, blue native polyacrylamide gel electrophoresis and immunoblotting
Mitochondria from cybrids were isolated as previously described.13 Individual Oxidative phosphorylation system (OXPHOS) complexes were extracted from mitochondria using n-dodecyl-β-D-maltoside (Sigma, St Louis, MO, USA) with a Detergent/protein ratio of 2.5 g per g. Protein (60 μg) containing 0.5% Blue G-250 (Sigma) and 5% glycerol were run on a 3–11% gradient blue native polyacrylamide gel electrophoresis as previously described.14 Immunoblotting of OXPHOS complexes was performed as described in another study,2 and blotting was done with anti-NDUFA13 (Grim19) (1:1000; MitoSciences, Eugene, OR, USA), anti-succinate dehydrogenase complex subunit A (SDHA) (1:1000; MitoSciences), anti-Core2 (1:1000; MitoSciences,), anti-cytochrome c oxidoreductase subunit I (COXI) (1:11000; MitoSciences) and anti-ATP synthase subunit alpha (1:11000; MitoSciences) antibodies. Anti-Lon (1:1000; Abcam, Bristol, UK, Anti-AFG3L2 (1:1000; Abcam), anti-ClpP/X (1:1000; Sigma), anti-HSP60 (1:1000; Cell Signaling, Danvers, MA, USA) and anti-GRP75 (1:1000; Santa Cruz, Dallas, TX, USA) were used to detect mitochondrial chaperone proteins. Anti-VDAC1 (1:1000; Cell Signaling) and anti-beta-Actin (1:1000; Santa Cruz) antibodies were served as internal control for mitochondria and whole cell extract respectively. Integrated optical density quantification was performed using a Gel-Pro Analyzer 4.0 (MediaCybernetics).
Respiratory complex enzymatic activity, and in-gel activity assay
For complex I in-gel activity assays, gels were soaked in assay buffer (25 mg nitrotetrazolium blue (Sigma), 1 μg ml−1 Nicotinamide adenine dinucleotide (NADH) (Sigma) and 5 mM Tris-HCL (pH 7.4)) for approximately 1 h at room temperature. The enzyme activity of four mitochondrial respiratory chain complexes were measured in the mitochondria of cybrids as described previously.15 The activity of each enzyme was normalized against that of VDAC, a protein located in mitochondrial out membrane.
Oxygen consumption
Endogenous oxygen consumption from intact cells was determined using a Oxygraph-2k (Oroboros, Innsbruck, Austria) as described previously.16 After recording the basal respiration, oligomycin (2.5 μg ml−1) (Sigma) was added to measure the phosphorylation-coupled respiration.
Statistical analysis
The data were presented as mean±s.d. from three independent experiments. Statistical significant was evaluated by independent two-tailed Student’s t test using SPSS 21.0 software (IBM, Armonk, NY, USA). A null hypothesis was rejected when P<0.05.
Results
Patient
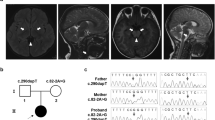
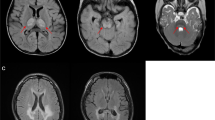
The patient (boy, aged 4 years at the time of this study) was born in a non-consanguineous Chinese family and presented with clinical manifestation of LS. The child was initially investigated for muscle weakness and psychomotor retardation at the age of 20 months. He was referred to our hospital at the age of 2 years and 2 months due to developmental delay. A first magnetic resonance imaging of the brain, performed at 9 months, showed a moderate cerebral atrophy and symmetric signal changes in basal ganglia (Figure 1a, red arrow in the left panel). A second magnetic resonance imaging, performed at the age of 1 year and 9 months, showed progression atrophy of the basal ganglia (Figure 1a, red arrow in the right panel). Physical examination indicated that the patient had masticatory atonia, limb hypotonia in general, muscle weakness with normal ankle clonus and positive Babinski sign. Although the patient growth was normal at his age of 20 months (weight 10 kg (normal 10.8–13.3 kg); height 85 cm (normal 81.9–88.4 kg); head circumference 45 cm (normal 44.2–52.1 cm)), reduced growth was found at the of 3 years and 4 months (weight 11 kg (normal 13.9–17.6 kg); height 93 cm (normal 95.0–103.1 cm); head circumference 47 cm (normal 46.2–53.9 cm)). Therefore, LS was considered in this patient.

magnetic resonance imaging of the brain, family tree, heteroplasmy and conservation analysis of the mutation. (a) Axial T2-weighted image of the brain of the patient, left panel showing high patch cords T2WI signal in bilateral globus pallidus and moderate cerebral atrophy. High T2WI signal was detected in the head of caudate nucleus and putamen (red arrow in the left panel). In the right panel, an enhanced T2WI signal (red arrow in the right panel) and progression atrophy in the head of caudate nucleus and putamen were observed compared with that of the left image. (b) Proband was indicated with an arrow; mutant loads for blood (B), urinary epithelium (U), oral mucosal epithelium cells (O) and skin fibroblasts (F) were determined by PCR-RFLP. Note: mutant loads in round brackets were determined by real time PCR. (c) Sequence diagram of blood and urinary epithelium cells from the patient and his mother. (d) PCR-RFLP analysis of MT-ND4 sequences of the patient (P), patient’s mother (M), patient’s aunt (A) and patient's grandma (G) using enzyme Rsa I, M: DNA marker. B: blood; U: urinary epithelium; O: oral mucosal epithelium cells; F: skin fibroblasts. All relatives are on the maternal side of the patient. (e) Conservation analysis showing the 11240C>T transition in the MT-ND4 gene (L161F substitution). L, Leu; F, Phe. A full color version of this figure is available at the Journal of Human Genetics journal online.
Though lactic acid (1.88 mmol l−1; normal range, 1.0–2.1 mmol l−1) and lactate/pyruvate ratio (14.2; normal range <20) were normal in the blood of the patient at the age of 12 months, they were increased to 2.7 mmol l−1 and 21.26 after 8 months, respectively. Following treatment with a combination of Levodopa and Benserazide Hydrochloride Tablet (30 mg per day), vitamin B1 (30 mg twice daily (b.i.d.)), vitamin B2 (2.5 mg b.i.d.), vitamin C (100 mg per day) and coenzyme Q10 (10 mg b.i.d.) at 2 years and 2 months of age, symptoms such as eyes movements and mental retardation improved significantly after 1 year of treatment.
Identification of pathogenic mutations
We performed targeted next-generation sequencing of MitoExome in this patient to uncover the genetic basis of LS. Based on the criteria raised by Sarah E. Calvo et al,10 none of the mitochondrial disease-related nuclear DNA mutation was detected in this patient. A novel mtDNA mutation of m.11240C>T was found in the heteroplasmic state in blood cells from patient (Table 2). The mutation was confirmed by Sanger sequencing in the blood and urinary epithelium cell from the patient (Figure 1c). On the contrary, such mutation was not detected in his mother using the method of Sanger sequencing (Figure 1c). We next screened the mutant load with the method of PCR-RFLP in the blood, oral mucosal epithelium cells and urinary epithelium cells from the patient and his healthy maternal family members. As shown in Figures 1b and d, mutant loads were higher in blood, urinary epitheliu, and oral mucosal epithelium cells of the patient compared with that of his asymptomatic mother (80.34 vs 21.16% in blood cells; 73.63 vs 27.08% in urinary epithelium cells; 71.21 vs 28.15% in oral mucosal epithelium cells). Furthermore, high mutant loads of m.11240C>T was confirmed in patient skin fibroblasts (66.7%). Such mutation was not detected in the blood from the maternal grandmother and the aunt on the mother side of the patient using the method of PCR-RFLP. To carefully determine the mutant loads of m.11240C>T in this family, a quantitative real-time PCR was applied. As shown in Figure 1b, mutant loads of m.11240C>T in the patient were 88.4% in the blood and 78.57% in the skin fibroblasts. Mutation of m.11240C>T was detectable in all maternal family members but with much lower mutant loads (24.64% in mother, 16.79% in aunt and 5.25% in grandmother) (Figure 1b). In addition, this mutation producing an amino acid change from leucine to phenylalanine in a highly conversed region of the ND4 protein (http://mtsnp.tmig.or.jp/) (Figure 1e) indicated a possible functional alteration of this mutation. Taken together, our findings reveal a likely pathogenic potential of the m.11240C>T mutation.
Construction of cybrids with m.11240C>T mutation
To fully understand the pathogenic role of m.11240C>T, we generated a series of cybrids with and without m.11240C>T mutation. Briefly, platelets from the patient were fused with mtDNA-less ρ0 human osteosarcoma 143B cells, and 16 single cybrids were selected by culturing the fusion mixtures in medium without uridine and sodium pyruvate for 2 weeks. One and two clones with homoplasmic m.11240C (C cells) and m.11240T (M1 and M2 cells), respectively, were selected by PCR-RFLP for further analysis (Figure 2a). In the meantime, mutation of m.11240C>T was confirmed in M1 and M2 clones by Sanger sequencing (Figure 2b). Wild-type 143B cells were also included to clearly understand the implications of m.11240C>T in mitochondrial dysfunction.

Cell models of m.11240C>T. (a) PCR-RFLP analysis of MT-ND4 sequences from C, M1 and M2 cells using enzyme Rsa I, M: DNA marker. (b) Sequence diagram of C, M1 and M2. WT: wild-type 143B cells; C: cells with m.11240C; M1 and M2: cells with m.11240T. A full color version of this figure is available at the Journal of Human Genetics journal online.
Blue native polyacrylamide gel electrophoresis analysis of respiratory complex assembly
To examine if the mutation of m.11240C>T in ND4 impaired the assembly of CI, the steady-state levels of complex I, complex II (succinate dehydrogenase), complex III (ubiquinol-cytochrome c reductase), complex IV (cytochrome c oxidase) and complex V (ATP synthase) of OXPHOS were determined by blue native polyacrylamide gel electrophoresis and immunoblotting. Mitochondria isolated from two control cells (WT and C) and two mutant cybrids (M1 and M2) were separated by blue native polyacrylamide gel electrophoresis for the analysis of in-gel complex I activity and immunoblotting analysis of the OXPHOS complexes. As shown in Figure 3a, the steady-state levels of all five OXPHOS complexes were not affected in the cybrids with homoplasmic m.11240C>T mutation compared with control cybrid without mutation. As the assembly of OXPHOS complexes were not affected in mutant cybrids, it was no surprise that the expression levels of quality control proteins such as Lon protease, GRP75, HSP60, AFG3L2, CLpP, and CLpX were not affected in mutant cybrids relative to that of control cells (Figure 3b). However, in-gel activity assay of complex I showed that the homoplasmic mutant cybrids of M1 and M2 exhibited a mildly reduced CI activity compared with that of two control cells (Figure 3c).

Respiratory complex assembly and in-gel activity assay. (a) Whole cells were solubilized with Triton X-100 (2%) and then subjected to BN-polyacrylamide gel electrophoresis (PAGE)/immunoblot analysis. Complexes I, II, III, IV and V were blotted with anti-Grim19, SDHA, Core2, COX1 and ATP-5A antibodies, respectively; VDAC was used as internal control. (b) The expression of LON, GRP75, HSP60, AFG3L2, CLPP, CLPX and ACTIN proteins in whole cells was examined by western blotting. (c) Mitochondria were solubilized with n-dodecyl-β-D-maltoside (2.5 g per g). In-gel activity assays of complex I was performed followed by BN-PAGE analysis; VDAC protein was used as internal control. C: m.11240C; M1 and M2: m.11240T; PC: muscle tissue from wild-type mouse. Results are representative of three independent experiments. Error bars,±s.d. *P<0.05. WT: wild-type 143B cells; C: cells with m.11240C; M1 and M2: cells with m.11240T. BN-PAGE, blue native polyacrylamide gel electrophoresis. A full color version of this figure is available at the Journal of Human Genetics journal online.
Functional analysis of mitochondrial respiratory chain
To further study the effect of m.11240C>T in the respiratory chain complexes, we measured respiratory chain complex activity in control and mutant 143B cells. As shown in Figure 4a, the mean activity levels of CI (60.89% decrease for M1 and 49.86% decrease for M2) and complex III (30.48% decrease for M1 and 20.43% decrease for M2) from mutant 143B cells were lower than those from control 143B cells. However, the activity of CII and CIV were similar between control and mutant 143B cells. Therefore, our results indicated that mutation of m.11240C>T impaired the activity of CI, which might affect the activity of CIII further. To fully understand the role of m.11240C>T on mitochondrial function, endogenous respiration of intact 143B cells were measured. As shown in Figure 4b, two mutant cybrids exhibited lower basal endogenous respiration and phosphorylation-coupled respiration than did control 143B cells. Thus, our findings confirmed that m.11240C>T impaired mitochondrial function by affecting the respirator activity of complex I.

Respiration assay of cells containing the m.11240C>T mutation. (a) Mitochondrial complexes enzyme activity of complex I, CII, CIII and CIV were measured in mitochondria isolated from control and mutant cells; VDAC protein was used as internal control. (b) Mitochondrial respiratory capacities were determined in control and mutant cells (n⩾3). Oligomycin (2.5 μg ml−1) was added for the measurement of uncoupled mitochondrial respiration (n⩾3). OXPHOS coupling respiration was calculated by subtracting the uncoupled component value from the total endogenous respiration value. Error bars,±s.d. *P<0.05. C: cells with m.11240C; M1 and M2: cells with m.11240T. A full color version of this figure is available at the Journal of Human Genetics journal online.
Discussion
By using MitoExome sequencing technology, we have identified a novel missense m.11240 C>T [p.Leu207Phe] mutation in the mitochondrial ND4 gene from a patient diagnosed with LS in the current study. To assess the pathogenicity of m.11240C>T mutation, we performed a systematic evaluation of this mutation at the clinical, genetic and molecular levels.
Clinically, the symptoms of eyes movements and mental retardation in the patient with a high level of m.11240C>T mutation was partially alleviated by the administration of a combination of supplements for the treatment of OXPHOS deficiency. Together with the fact that the mutant loads of m.11240C>T was much higher in patient than that in his mother, and other maternal relatives. This led us to believe that m.11240C>T may play an important role in the disease pathogenesis.
Genetically, the m.11240C>T lead to a leucine to phenylalanine replacement at a highly conversed codon region of ND4 gene. This mtDNA variant was not found in any of the published mtDNA polymorphism databases from MITOMAP (http://www.mitomap.org), mtDB (http://www.genpat.uu.se/mtDB/), mtSNP (http://mtsnp.tmig.or.jp) and Google. Additionally, we found that m.11240C>T was not detected in our private control database with more than 500 local healthy subjects.2
At the molecular level, cybrids with m.11240C>T exhibited a lower mitochondrial complex I activity than did m.11240C and 143B wild-type cells in two different experiments (in-gel activity assay and enzyme activity assay). Although in-gel activity of complex I was found not to be affected by ND4 mutation, our results confirmed that mutation of ND4 impaired the activity of peripheral arm of complex I.17, 18 To be noted, by blue native polyacrylamide gel electrophoresis analysis on ND4 mutant cybrids, we found the complex I assembly was normal when compared with both control cybrids and 143B wild-type cells. Given the fact that ND4 is one of the proton pumping channels of complex I.19 It is possible that the mutation of m.11240C>T in the fourth hydrophobic trans-membrane region has little effect on the complex I assembly (http://www.uniprot.org/uniprot/P03905). Alternatively, the proton pumping channel of complex I, which was composed by 11 trans-membrane regions of ND4, was more or less impaired due to the mutation in one trans-membrane region. Similar phenomenon was found in a patient with m.13063G>A in ND5 in a previous report.20 Since ND5 is not a ubiquinone binding subunit19 and the mutation of m.13063G>A is located in the seventh trans-membrane region, we speculated that without affecting the assembly of holoenzyme, the trans-membrane region of ND5 was probably impaired to block the proton pumping as we found in ND4.
Mutations in ND4 were frequently observed in patients with mitochondrial disorders such as Leber hereditary optic neuropathy,21 LS22 and chronic progressive external ophthalmoplegia.23 In this study, we reported a novel m.11240C>T mutation in ND4 gene that was associated with LS with functional evidence. Until recently, there were a total of 15 ND4 mutations reported in mitomap (www.mitomap.org)24; of these, only four of them, including m.11778G>A,25 m.11696G>A,26 m.11777G>A22 and m.11832G>A,27 had functional studies available. m.11232T>C and m.11253T>C are two mitochondrial diseases-related point mutations that close to m.11240C>T; however, underlying mechanisms of these two mutations in mitochondrial diseases were not known. As both m.11232T>C and m.11253T>C are located in the same trans-membrane region of m.11240C>T, we believe that these two mutations might share the same mechanism of complex I defect as m.11240C>T did.
In summary, we report here that m.11240C>T as a new missense mutation in the ND4 gene that was associated with LS. Mutation of m.11240C>T impaired mitochondrial respiration by decreasing the activity of mitochondrial complex I.
References
Nouws, J., Nijtmans, L. G., Smeitink, J. A. & Vogel, R. O. Assembly factors as a new class of disease genes for mitochondrial complex I deficiency: cause, pathology and treatment options. Brain 135, 12–22 (2012).
Fang, H., Shi, H., Li, X., Sun, D., Li, F., Li, B. et al. Exercise intolerance and developmental delay associated with a novel mitochondrial ND5 mutation. Scientific rep. 5, 10480 (2015).
Ferreira, M., Torraco, A., Rizza, T., Fattori, F., Meschini, M. C., Castana, C. et al. Progressive cavitating leukoencephalopathy associated with respiratory chain complex I deficiency and a novel mutation in NDUFS1. Neurogenetics 12, 9–17 (2011).
Baracca, A., Solaini, G., Sgarbi, G., Lenaz, G., Baruzzi, A., Schapira, A. H. et al. Severe impairment of complex I-driven adenosine triphosphate synthesis in leber hereditary optic neuropathy cybrids. Arch. Neurol. 62, 730–736 (2005).
Liolitsa, D., Rahman, S., Benton, S., Carr, L. J. & Hanna, M. G. Is the mitochondrial complex I ND5 gene a hot-spot for MELAS causing mutations? Ann. neurol. 53, 128–132 (2003).
Loeffen, J., Elpeleg, O., Smeitink, J., Smeets, R., Stockler-Ipsiroglu, S., Mandel, H. et al. Mutations in the complex I NDUFS2 gene of patients with cardiomyopathy and encephalomyopathy. Ann. neurol. 49, 195–201 (2001).
Lightowlers, R. N., Taylor, R. W. & Turnbull, D. M. Mutations causing mitochondrial disease: what is new and what challenges remain? Science 349, 1494–1499 (2015).
Finsterer, J. Leigh and Leigh-like syndrome in children and adults. Pediatr. Neurol. 39, 223–235 (2008).
Cui, H., Li, F., Chen, D., Wang, G., Truong, C. K., Enns, G. M. et al. Comprehensive next-generation sequence analyses of the entire mitochondrial genome reveal new insights into the molecular diagnosis of mitochondrial DNA disorders. Genet. med. 15, 388–394 (2013).
Calvo, S. E., Compton, A. G., Hershman, S. G., Lim, S. C., Lieber, D. S., Tucker, E. J. et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci. Transl. Med. 4, 118ra110 (2012).
Chomyn, A., Lai, S. T., Shakeley, R., Bresolin, N., Scarlato, G. & Attardi, G. Platelet-mediated transformation of mtDNA-less human cells: analysis of phenotypic variability among clones from normal individuals—and complementation behavior of the tRNALys mutation causing myoclonic epilepsy and ragged red fibers. Am. j. hum. genet. 54, 966–974 (1994).
Sambrook, J. & Russell, D. W. Molecular cloning: a laboratory manual, (Cold Spring Harbor Laboratory Press, New York, 2001).
Fernandez-Vizarra, E., Ferrin, G., Perez-Martos, A., Fernandez-Silva, P., Zeviani, M. & Enriquez, J. A. Isolation of mitochondria for biogenetical studies: an update. Mitochondrion 10, 253–262 (2010).
Wittig, I., Braun, H. P. & Schagger, H. Blue native PAGE. Nat. protoc. 1, 418–428 (2006).
Birch-Machin, M. A. & Turnbull, D. M. Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods Cell Biol. 65, 97–117 (2001).
Bai, Y. & Attardi, G. The mtDNA-encoded ND6 subunit of mitochondrial NADH dehydrogenase is essential for the assembly of the membrane arm and the respiratory function of the enzyme. EMBO j. 17, 4848–4858 (1998).
Pello, R., Martin, M. A., Carelli, V., Nijtmans, L. G., Achilli, A., Pala, M. et al. Mitochondrial DNA background modulates the assembly kinetics of OXPHOS complexes in a cellular model of mitochondrial disease. Hum. Mol. Genet. 17, 4001–4011 (2008).
Jiang, P., Liang, M., Zhang, J., Gao, Y., He, Z., Yu, H. et al. Prevalence of mitochondrial ND4 mutations in 1281 Han Chinese subjects with Leber's hereditary optic neuropathy. Invest. Ophthalmol. Vis. Sci. 56, 4778–4788 (2015).
Sazanov, L. A. A giant molecular proton pump: structure and mechanism of respiratory complex I. Nat. Rev. Mol. Cell Biol. 16, 375–388 (2015).
Malfatti, E., Bugiani, M., Invernizzi, F., de Souza, C. F., Farina, L., Carrara, F. et al. Novel mutations of ND genes in complex I deficiency associated with mitochondrial encephalopathy. Brain 130, 1894–1904 (2007).
Bolhuis, P. A., Bleeker-Wagemakers, E. M., Ponne, N. J., Van Schooneveld, M. J., Westerveld, A., Van den Bogert, C. et al. Rapid shift in genotype of human mitochondrial DNA in a family with Leber's hereditary optic neuropathy. Biochem. Biophys. Res. Commun. 170, 994–997 (1990).
Komaki, H., Akanuma, J., Iwata, H., Takahashi, T., Mashima, Y., Nonaka, I. et al. A novel mtDNA C11777A mutation in Leigh syndrome. Mitochondrion 2, 293–304 (2003).
Pulkes, T., Liolitsa, D., Nelson, I. P. & Hanna, M. G. Classical mitochondrial phenotypes without mtDNA mutations: the possible role of nuclear genes. Neurology 61, 1144–1147 (2003).
Ruiz-Pesini, E., Lott, M. T., Procaccio, V., Poole, J. C., Brandon, M. C., Mishmar, D. et al. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res. 35, D823–D828 (2007).
Hofhaus, G., Johns, D. R., Hurko, O., Attardi, G. & Chomyn, A. Respiration and growth defects in transmitochondrial cell lines carrying the 11778 mutation associated with Leber's hereditary optic neuropathy. J. Biol. Chem. 271, 13155–13161 (1996).
De Vries, D. D., Went, L. N., Bruyn, G. W., Scholte, H. R., Hofstra, R. M., Bolhuis, P. A. et al. Genetic and biochemical impairment of mitochondrial complex I activity in a family with Leber hereditary optic neuropathy and hereditary spastic dystonia. Am. J. Hum. Genet. 58, 703–711 (1996).
Taivassalo, T., Shoubridge, E. A., Chen, J., Kennaway, N. G., DiMauro, S., Arnold, D. L. et al. Aerobic conditioning in patients with mitochondrial myopathies: physiological, biochemical, and genetic effects. Ann. neurol. 50, 133–141 (2001).
Acknowledgements
This work was supported by the Zhejiang Provincial Natural Science foundation of China (LQ15H070005), Key Science and Technology Innovation Team of Zhejiang Province (2010R50048), Chinese National Science Foundation (81471097) and the Start Foundation of Wenzhou Medical University (to Hezhi Fang).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Xu, B., Li, X., Du, M. et al. Novel mutation of ND4 gene identified by targeted next-generation sequencing in patient with Leigh syndrome. J Hum Genet 62, 291–297 (2017). https://doi.org/10.1038/jhg.2016.127
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2016.127
This article is cited by
-
A Novel NDUFS3 mutation in a Chinese patient with severe Leigh syndrome
Journal of Human Genetics (2018)
