
Abstract
Osteogenesis imperfecta (OI) is a group of hereditary disorders characterized by decreased bone mass and increased fracture risk. The majority of OI cases have an autosomal dominant pattern of inheritance and are usually caused by mutations in genes encoding type I collagen. OI cases of autosomal recessive inheritance are rare, and OI type XI is attributable to mutation of the FKBP10 gene. Here, we used next-generation sequencing and Sanger sequencing to detect mutations in FKBP10 and to analyze their relation to the phenotypes of OI type XI in three Chinese patients. We also evaluated the efficacy of zoledronic acid treatment in these patients. Two of the affected patients had novel compound heterozygous mutations, one patient with c.343C>T (p.R115X) in exon 2 and c.1085delC (p.A362fsX1) in exon 7, and the other patient with c.879C>G (p.Y293X) in exon 5 and c.918-3C>G in intron 5. In the third proband, we identified a homozygous single base-pair duplication, c.831dupC (p.G278RfsX95) in exon 5. In conclusion, we report for the first time that these novel pathogenic mutations of FKBP10 can lead to the extremely rare type XI OI without contractures, which expands the genotypic spectrum of OI. The phenotypes of these patients are similar to patients with types III or IV OI, and zoledronic acid is effective in increasing BMD, inhibiting bone resorption biomarkers and reducing fractures of these patients.
Similar content being viewed by others

Introduction
Osteogenesis imperfecta (OI), also known as ‘brittle bone disease’, is one of the most common heritable skeletal dysplasias and is characterized by bone fragility, low bone mass, recurrent fractures and bone deformity. Extra-skeletal features of OI include blue sclera, deafness and growth deficiency. Patients with OI have significant variable expressivity ranging from mild to neonatal lethality. The majority of OI cases (85–90%) have an autosomal dominant inheritance pattern and are usually caused by mutations in genes encoding type I collagen chains (COL1A1/COL1A2) or interferon-induced transmembrane protein 5 (IFITM5).1 These mutations lead to differences in the quantity or structure of type I collagen, or to impairments in osteoblast maturation.1, 2 Recent studies have identified mutations in several genes that give rise to autosomal recessive OI, including CRTAP, P3H1, PPIB, SERPINF1, SERPINH1, SP7, BMP1, TMEM38B, WNT1 and SPARC. These genes encode proteins involved in posttranslational modification, folding, secretion or processing of type I collagen, as well as in osteoblast differentiation and function.1, 3, 4, 5 The gene FKBP10 (17q21.2; MIM 607063) encodes FK506 binding protein (FKBP65), and mutation of FKBP10 would produce an extremely rare autosomal recessive form of OI.6, 7
FKBP65 is a 582-amino-acid glycoprotein that belongs to the highly conserved family of FKBPs with prolyl cis/trans isomerase (PPIase) activity. The endoplasmic reticulum resident FKBP65 is known to be involved in elastin binding/isomerization, and has demonstrated the ability to interact with triple helical collagen and function as a chaperone.8 In 2010, loss-of-function mutations in FKBP10 were firstly reported to cause autosomal recessive OI type XI (OMIM 610908) in families with progressive deforming of bones.7 Phenotypic features of OI type XI include severe osteoporosis, long bone fractures, ligamentous laxity, platyspondyly and scoliosis. Dentinogenesis imperfecta, hearing impairment and blue sclera are usually absent.7, 9, 10, 11 Histological assessment of bone revealed sparse and distorted lamellar structure.7, 12, 13 Patients with FKBP10 mutations can be classified as isolated OI, Bruck syndrome (MIM 259450/609220, with both congenital large joint contracture and bone fragility), or Kuskokwim syndrome (MIM 208200, with congenital contractures, but no major skeletal defects).10, 14, 15 We have previously reported FKBP10 mutations leading to rare Bruck syndrome.14 However, no information about the FKBP10 mutation and its isolated OI phenotypes is available in the Chinese population.
Here, we investigate the phenotypes of three Chinese patients with moderate-to-severe OI, detect mutations of the FKBP10 gene, and evaluate the effects of bisphosphonates (BPs) in these patients.
Materials and methods
Subjects
Three Chinese patients from different non-consanguineous families were clinically diagnosed with OI in the Department of Endocrinology of Peking Union Medical College Hospital (PUMCH) between 2009 and 2013. The study was approved by the Ethics Committee of PUMCH. Signatures for informed consent were obtained from the patients and their parents before their participation in the study.
Phenotypes evaluation
A detailed medical history was collected and physical examination was completed. Fracture count was determined by medical history and bone X-rays. Age- and sex-adjusted height and weight were calculated based on the standardized growth charts for Chinese children.16 Mobility was characterized by the brief assessment of motor function (BAMF, 10-point ordinal scale), a rapid evaluation of gross motor performance and proved for reliability in OI population.17 Bone mineral density (BMD) at lumbar spine 2–4 (LS) and femoral neck (FN) was measured by dual-energy X-ray absorptiometry (DXA, Lunar Prodigy, GE Healthcare, USA). BMD Z-scores were calculated based on data from age matched normal children in China.18, 19, 20 Serum levels of calcium (Ca), phosphate (P), alanine aminotransferase (ALT) and creatinine (Cr) were measured by automatic analyzer. Serum concentrations of β-cross-linked C-telopeptide of type I collagen (β-CTX, bone resorption marker), total alkaline phosphatase (bone formation marker), 25 hydroxyvitamin D (25OHD, marker of vitamin D nutritional status) and intact parathyroid hormone were measured using an automated electrochemiluminescence system (E170; Roche Diagnostics, Switzerland). Serum bone specific alkaline phosphatase (BALP, bone formation marker) was determined with an IRMA kit (OSTASE, Beckman Coulter, Brea, CA, USA), and the levels were compared with age/sex-specific references in healthy children.21 All tests were performed in the clinical central laboratory of PUMCH.
The three patients received intravenous infusion with zoledronic acid annually (5 mg; Aclasta, Novartis Pharma Stein AG, Basel, Switzerland). Treatment efficacy was evaluated by the changes in BMD, bone turnover biomarkers, fracture incidence and mobility scores from baseline. Safety was assessed by adverse effects of zoledronic acid and biomarkers of liver and renal functions.
Pathogenic mutation detection
Next-generation sequencing
Genomic DNA of the probands and their parents was extracted from peripheral leukocytes with a QIAamp DNA Mini Kit (50) (Qiagen, Germany). A customized next-generation sequencing panel (Illumina HiSeq2000 platform, Illumina, Inc., San Diego, CA, USA) was used to capture all exons, splice sites and immediate flanking intron sequences of 700 genes involved in skeletal disorders, including the causative genes of OI: COL1A1, COL1A2, IFITM5, SERPINF1, CRTAP, P3H1, PPIB, SERPINH1, FKBP10, BMP1, SP7, WNT1, PLOD2 and TMEM38B. The overall sequencing coverage of the target regions was 98.95% for 200 × depth of coverage in each of the chromosomes.
Analysis of variants was restricted to coding regions and flanking intronic sequences. The resulting variants were filtered using SAMtools (version 1.4) and SOAPsnp software (version 2.0) to exclude variants with a minor allele frequency ⩾0.005 in the Single Nucleotide Polymorphism Database (dbSNP build 137), the Exome Variant Server, the 1000 Genomes Project, the National Heart, Lung and Blood Institute (NHLBI), or the UCSC common single nucleotide polymorphism database and internal control database.22, 23 In our study, subsequent analysis depended on the clinical diagnosis of OI: variants in genes causing recessive OI were considered to be pathogenic if they were homozygous or compound heterozygous; and mutations leading to premature chain termination (frameshift and nonsense) or affecting splice sites were considered pathogenic (Supplementary Table S1).23
Sanger sequencing
PCR and Sanger sequencing was used to confirm next-generation sequencing variants and provide quality assurance. According to the results of next-generation sequencing, the corresponding fragments of FKBP10 gene were amplified by PCR. Primers were designed using Primer3 (http://www.sourceforge.net;Supplementary Table S2). PCR was performed with the following protocol: initial denaturation at 95 °C for 3 min, followed by 35 cycles of 95 °C for 30 s, 59–61 °C for 30 s and 72 °C for 60 s. Direct sequencing of PCR products were performed on an ABI 3130 automatic sequencer (Applied Biosystems, Foster City, CA, USA) using standard methods. Sanger sequencing were also performed for 100 unrelated, healthy and ethnically matched subjects to confirm that the mutations were not merely polymorphisms. The results of sequencing were compared with the reference sequence of FKBP10 (NM_021939.3).
Reverse transcription-PCR
To evaluate the changes of messenger RNA induced by the splicing mutation, reverse transcriptase-PCR amplification was performed. Total RNA from whole blood was isolated using the RNA Extraction Kit (Qiagen, Germany). The complementary DNA was then generated from total RNA by reverse transcriptase-PCR reaction using the Goscript Reverse Transcription system kit (Promega, Madison, WI, USA). Specific primer for PCR amplification of FKBP10 complementary DNA was designed (forward 5′-TGGATGTGTGGAACAAGGAA-3′; reverse 5′-GGTGGTCTCATTGCAGGTCT-3′). PCR was performed using the methods similar to above mentioned.
Results
Clinical presentation
Patients in all three families shared typical features of OI without joint contractures. Family histories were negative for bone disorders. Clinical findings are summarized in Table 1.
Family 1
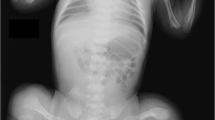
The proband, a 20-year-old girl, was born full term by cesarean delivery with a weight of 2600 g (5th centile). She was the only affected daughter of non-consanguineous parents. Her first brittle fracture occurred at 52 days after birth. Subsequently, nearly sixty fractures occurred under minor trauma at her bilateral lower limbs and left humerus. By the age of 16 years, her movement was greatly restricted and she was able to ambulate only by wheelchair. Physical examination indicated short stature, low weight (110 cm and 35 kg; an average of 5 year and 11-year-old girl at chronological age of 16 years, respectively), scoliosis, apparent bending deformities of limbs and joint hyperextensibility (Figure 1). She had normal sclera, hearing, dentition and intelligence. X-ray revealed severe osteoporosis with occipital wormian bone, multiple vertebral compression fractures with scoliosis, collapsed thoracic cage, deformities of upper and lower limbs, slender long bone with reduced cortical thickness, and popcorn formation (Figure 1). She received intravenous infusion of zoledronic acid annually since she was 16 years old.

Phenotypes of the three osteogenesis imperfecta patients with FKBP10 mutations. Physical findings: for patient 1, (a, b) short stature, severe bending deformities of lower and left upper limbs (white arrows); for patient 2, (c, d) faint blue sclera and normal teeth, (e) deformity of the left lower limb (white arrow) without contractures, (f, g) sternum protrusion (white arrow) with no ithyokyphosis. Radiological findings: for patient 1, (h) deformity of the left upper limb, (i, j) severe osteoporosis with old fracture (white arrow), deformities of pelvis and bilateral lower limbs, slender long bone with reduced cortical thickness and popcorn formation (white arrows), (k, l) vertebral compression fractures (white arrows), scoliosis (white arrow), indistinct ribs and thoracic cage collapse (white arrows); for patient 2, (m) slender long bone with thin cortices, bowing femora (white arrows) with healed fractures (white arrow), (n) occipital wormian bones (white arrow), (o) no ithyokyphosis; for patient 3, (p) severe osteoporosis, and gracile femora with intramedullary rodding (white arrows). A full color version of this figure is available at the Journal of Human Genetics journal online.
Family 2
The proband, a 5-year-old boy, was born full term by normal delivery with a birth weight of 3100 g (25th centile). He was the second child of a non-consanguineous couple. Since the age of 2 years, he experienced recurrent bilateral femoral, left tibia and left fibula fractures. At 4 years old, he presented with failure to thrive and was unable to ambulate. Intelligence and other developmental milestones were normal. Physical examination revealed short stature and low weight (80 cm and 13 kg; an average of 15 month and 2-year-old boy at chronological age of 4 years, respectively), faint blue sclera, deformities of left lower limb and sternum protrusion (Figure 1). He had no hearing loss, dentingenesis imperfecta or joint hyperextensibility. X-ray films revealed severe osteoporosis with occipital wormian bone, slender long bone with thin cortices, and bowing femora with healed fractures (Figure 1). Treatment with intravenous zoledronic acid was started at 4 years of age.
Family 3
The patient, a 9-year-old girl, was born at term with a birth weight of 3400 g (50th centile). She was the only child of a non-consanguineous couple. She incurred multiple bone fractures at her right humerus, caudal vertebra and bilateral femora under trivial trauma when she was 2, 4, and 5 years old. At 5 years old, she was still unable to stand and walk. Physical examination revealed her stature was at the median of age- and sex-adjusted children level. She had joint hyperextensibility and costal eversion. X-ray films indicated severe osteoporosis, multiple wormian bones and gracile femora with thin cortices (Figure 1). Treatment with intravenous zoledronic acid was started at 5 years of age.
Mutations of FKBP10
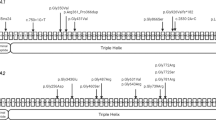
For patient 1, compound heterozygous mutations in FKBP10 were identified: a c.343C>T in exon 2 leading to generation of a premature termination codon (p.R115X), and a c.1085delC in exon 7 leading to a translational frameshift (p.A362fsX1; Supplementary Figure S1). The patient 2 also harbored compound heterozygous mutations of FKBP10: a c.879C>G in exon 5 leading to a premature termination codon (p.Y293X) and a c.918-3C>G located three bases upstream of exon 6 (Supplementary Figure S1). For the evaluation of splicing mutation, we performed reverse transcription-PCR but failed to amplify FKBP10 messenger RNA from peripheral blood of both the patient and healthy controls. Parents of the two patients were heterozygous carriers of the compound mutations. For patient 3, a homozygous duplication (c.831dupC) in exon 5 of FKBP10 was identified (Supplementary Figure S1). The mutation led to a premature termination codon only 95 amino acids downstream (p.G278RfsX95). The parents were heterozygous carriers of this mutation, implying a hidden consanguinity or a founder mutation in the population of origin.
The four mutations in FKBP10 in family 1 and 2 are novel and absent from 100 healthy controls, while the mutation in family 3 has been previously identified.7, 10, 11, 12, 24 No mutations were found in other genes related to OI.
Efficacy of zoledronic acid therapy
At baseline, serum Ca, P, total alkaline phosphatase and BALP concentrations were within age-appropriate normal ranges, whereas bone resorption marker β-CTX levels were higher than normal range. BMD and its Z-scores at LS and FN were extremely low (Table 1).
The first patient was treated with zoledronic acid annually for 5 years, and BMD significantly increased by +2.2 s.d. and +4.2 s.d. at LS and FN, respectively (Table 1, Figure 2), with only one new fracture occurring at the left limb during the follow-up. The second patient was treated with zoledronic acid annually for 2 years. Serum β-CTX level decreased after 24-month treatment, and BMD at LS and FN significantly increased by +6.1 s.d. and +4.7 s.d., respectively (Table 1,Figure 2). One new fracture occurred at the left femur after 9 months of zoledronic acid treatment. The third patient suffered a right femoral fracture at the third month after the first infusion of zoledronic acid, which results in the suspending of BPs for almost 2 years. The fracture eventually healed after a surgery with intramedullary rodding when the patient was 8 years old, and subsequently she restarted the therapy annually for 2 years. At the most recent assessment, BMD at LS significantly increased by +3.6 s.d., and FN BMD were not calculated due to intramedullary rodding (Table 1, Figure 2). After treatment, no new vertebral compression fractures were observed when we compared the X-ray films of pretreatment and the last follow-up. Moreover, the movement abilities were improved after treatment: patient 1 was able to sit freely without any support (+1 BAMF score); patient 2 started walking with no gait device (+3 BAMF score); and patient 3 could ambulate with a walker support (+3 BAMF score; Table 1). Three patients had mild fever within the first three days after the first infusion of zoledronic acid. No other adverse events were observed.

Effects of zoledronic acid on BMD and Z-scores in the three patients with osteogenesis imperfecta. (a) BMD and (b) its Z-scores at LS, FN were significantly increased during treatment with zoledronic acid in the three patients. For the patient 3, the false elevated of FN BMD and Z-scores due to intramedullary rodding were not shown in a, b. BMD, bone mineral density; FN, femoral neck; LS, lumbar spine 2–4; ZOL, zoledronic acid. A full color version of this figure is available at the Journal of Human Genetics journal online.
Discussion
OI type XI is an extremely rare autosomal recessive, moderate-to-severe form of OI that is attributed to mutations of the FKBP10 gene. We report for the first time that mutations in FKBP10 lead to isolated OI without contractures in Chinese population, which expand the phenotypic range of mutations in FKBP10. The causative mutations of the three patients include four novel compound heterozygous mutations and a known homozygous mutation in FKBP10. Three patients presented with severe osteoporosis, early-onset recurrent fractures, progressive bone deformities and growth deficiency. Intravenous zoledronic acid is effective to increase BMD, reduce fracture incidence and improve mobility in patients with FKBP10 mutations.
Until now, 29 unique sequence variants have been identified in FKBP10 gene (Figure 3; http://lovd.nl/fkbp10). The homozygous mutation of c.831dupC we identified in patient 3 has been reported previously in 13 patients of Mexican, Turkish, Pakistan, South African or Caucasian origin.7, 10, 11, 12, 24 In contrast, we identified the compound heterozygous mutations of c.343C>T in exon 2, c.879C>G in exon 5, c.918-3C>G in intron 5 and c.1085delC in exon 7 of FKBP10, which are all novel pathogenic mutations of OI. The encoding FKBP65 protein can prevent premature cross-links between procollagen chains, assist proper folding of collagen helix and promote subsequent trafficking.8, 13, 25 It is reported the postnatal FKBP10 expression is restricted to developing bone and tendons/ligaments.26 This suggests FKBP10 null mutations may promote bone fragility or joint contracture, roles consistent with the phenotypes in our patients. Inactivating mutations in FKBP10 may impair FKBP65 function, delay collagen secretion, diminish telopeptide hydroxylation, and then lead to reduced collagen cross-linking and deposition in extracellular matrix.12, 13, 27 In the present study, the nonsense or frameshift mutations in FKBP10 affect the important functional enzyme, nonsense-mediated messenger RNA decay will ensue and no truncated protein will be produced.28 Another mutation in a splice site (c.918-3C>G) may likely introduce alternatively spliced transcripts, but future reverse transcriptase PCR performed with fibroblasts is worthwhile.27 Finally, absence of FKBP65 can be expected to reduce the proper folding, secretion and cross-linking of type I procollagen.

Location of the novel and known FKBP10 variants in DNA and protein level. (a) Arrows indicate the sites of mutations of FKBP10 on the exons or exon–intron junctions. Blue boxes indicate the 10 exons of FKBP10. (b) Representation of FKBP65 with the locations of mutations in patients 1–3. The novel and known FKBP10 variants identified in this study are indicated in red and blue, respectively. A full color version of this figure is available at the Journal of Human Genetics journal online.
The phenotypes of Chinese patients with FKBP10 mutations are similar to those of patients with OI type XI (Table 2). The patients have moderate-to-severe phenotypes resembling OI type III–IV. We find that levels of the biomarker β-CTX are elevated, but otherwise find no alterations in the biomarkers total alkaline phosphatase, BALP or Ca (Table 1). FKBP10 mutation can cause either OI or Bruck syndrome, consistent with our earlier observations.14 Our third patient has OI without contractures, although she has the same recurrent mutation of FKBP10 (c.831dupC and p.G278RfsX95) as Bruck syndrome patients do.10, 11, 29 These findings indicate that mutations in FKBP10 lead to a large variability in phenotypes, ranging from isolated OI to Bruck syndrome. This striking phenotypic diversity distinguishes FKBP10 mutations from other causes of recessive OI. The position of the mutation within FKBP10 gene is not a good predictor of the severity of OI.
BPs therapy is demonstrated to markedly increase BMD, reshape compressed vertebral bodies, reduce fracture incidence and substantially improve functional status in children with OI.30 However, there is considerably less information concerning the effects of BPs in patients with FKBP10 mutations. In our patients, treatment with zoledronic acid led to a constant increase in BMD at the LS and proximal hip, reduction in long bone fracture incidence and improvement in ability of movement. In an Italy boy with a c.1399+1G>A splice mutation in FKBP10, higher BMD and fewer fractures are also observed after treatment with neridronate.27 Furthermore, a recent study in five patients with Bruck syndrome (one with FKBP10 mutation and four with PLOD2 mutation) indicates that the efficacy of zoledronic acid were comparable between Bruck syndrome and other OI patients.31 During our observation, we closely monitor the hepatic, renal function and find that zoledronic acid is well tolerated in our patients. Indeed the efficacy and safety of BPs therapy in patients with FKBP10 mutations still remains to be confirmed in large sample clinical studies over long period. Moreover, we notice our patients are no longer even osteopenic after 2–5 yrs of BPs therapy. The relative normal BMD Z-scores may support alternating treatment with periodic drug holidays to avoid over-treatment. However, there is no consensus on how long BPs treatment can be continued in OI.30, 32 More clinical data is needed to define an optimal duration to maximize fracture protection in patients with FKBP10 mutations.
The present study is limited by an in-depth investigation of the splicing defect in our second patient. In future, we should complete the functional study about the mutant FKBP65 in cultured fibroblasts, to reveal the underlying mechanism of c.918-3C>G leading to OI.
In conclusion, OI type XI is a rare autosomal recessive type of OI, which leads to multiple fractures, progressive bone deformities and growth deficiency. We identify four novel mutations in the FKBP10 gene (c.343C>T, c.879C>G, c.918-3C>G and c.1085delC) that lead to OI type XI without contractures, which expand the genotypic spectrum of OI. Zoledronic acid is demonstrated to be effective in increasing BMD, reducing fracture incidence and improving movement ability in patients with OI type XI.
References
Forlino, A. & Marini, J. C. Osteogenesis imperfecta. Lancet 387, 1657–1671 (2016).
Marini, J. C. & Blissett, A. R. New genes in bone development: what's new in osteogenesis imperfecta. J. Clin. Endocrinol. Metab. 98, 3095–3103 (2013).
Rohrbach, M. & Giunta, C. Recessive osteogenesis imperfecta: clinical, radiological, and molecular findings. Am. J. Med. Genet. C Semin. Med. Genet. 160C, 175–189 (2012).
Byers, P. H. & Pyott, S. M. Recessively inherited forms of osteogenesis imperfecta. Annu. Rev. Genet. 46, 475–497 (2012).
Mendoza-Londono, R., Fahiminiya, S., Majewski, J., Tetreault, M., Nadaf, J., Kannu, P. et al. Recessive osteogenesis imperfecta caused by missense mutations in SPARC. Am. J. Hum. Genet. 96, 979–985 (2015).
Coss, M. C., Winterstein, D., Sowder, R. C. & Simek, S. L. Molecular cloning, DNA sequence analysis, and biochemical characterization of a novel 65-kDa FK506-binding protein (FKBP65). J. Biol. Chem. 270, 29336–29341 (1995).
Alanay, Y., Avaygan, H., Camacho, N., Utine, G. E., Boduroglu, K., Aktas, D. et al. Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am. J. Hum. Genet. 86, 551–559 (2010).
Ishikawa, Y., Vranka, J., Wirz, J., Nagata, K. & Bachinger, H. P. The rough endoplasmic reticulum-resident FK506-binding protein FKBP65 is a molecular chaperone that interacts with collagens. J. Biol. Chem. 283, 31584–31590 (2008).
Steinlein, O. K., Aichinger, E., Trucks, H. & Sander, T. Mutations in FKBP10 can cause a severe form of isolated Osteogenesis imperfecta. BMC Med. Genet. 12, 152 (2011).
Kelley, B. P., Malfait, F., Bonafe, L., Baldridge, D., Homan, E., Symoens, S. et al. Mutations in FKBP10 cause recessive osteogenesis imperfecta and Bruck syndrome. J. Bone Miner. Res. 26, 666–672 (2011).
Shaheen, R., Al-Owain, M., Faqeih, E., Al-Hashmi, N., Awaji, A., Al-Zayed, Z. et al. Mutations in FKBP10 cause both Bruck syndrome and isolated osteogenesis imperfecta in humans. Am. J. Med. Genet. A 155A, 1448–1452 (2011).
Schwarze, U., Cundy, T., Pyott, S. M., Christiansen, H. E., Hegde, M. R., Bank, R. A. et al. Mutations in FKBP10, which result in Bruck syndrome and recessive forms of osteogenesis imperfecta, inhibit the hydroxylation of telopeptide lysines in bone collagen. Hum. Mol. Genet. 22, 1–17 (2013).
Barnes, A. M., Cabral, W. A., Weis, M., Makareeva, E., Mertz, E. L., Leikin, S. et al. Absence of FKBP10 in recessive type XI osteogenesis imperfecta leads to diminished collagen cross-linking and reduced collagen deposition in extracellular matrix. Hum. Mutat. 33, 1589–1598 (2012).
Zhou, P., Liu, Y., Lv, F., Nie, M., Jiang, Y., Wang, O. et al. Novel mutations in FKBP10 and PLOD2 cause rare Bruck syndrome in Chinese patients. PLoS ONE 9, e107594 (2014).
Barnes, A. M., Duncan, G., Weis, M., Paton, W., Cabral, W. A., Mertz, E. L. et al. Kuskokwim syndrome, a recessive congenital contracture disorder, extends the phenotype of FKBP10 mutations. Hum. Mutat. 34, 1279–1288 (2013).
Li, H., Ji, C. Y., Zong, X. N. & Zhang, Y. Q. Height and weight standardized growth charts for Chinese children and adolescents aged 0 to 18 years. Chin. J. Pediatr. 47, 487–492 (2009).
Cintas, H. L., Siegel, K. L., Furst, G. P. & Gerber, L. H. Brief assessment of motor function: reliability and concurrent validity of the Gross Motor Scale. Am. J. Phys. Med. Rehabil. 82, 33–41 (2003).
Genant, H. K., Grampp, S., Gluer, C. C., Faulkner, K. G., Jergas, M., Engelke, K. et al. Universal standardization for dual x-ray absorptiometry: patient and phantom cross-calibration results. J. Bone Miner. Res. 9, 1503–1514 (1994).
Zhang, L. W., Liu, J. C., Zhai, F. Y., Cao, R. X. & Duan, J. L. Normal reference values for bone mineral density in children and adolescents aged 6–18 years, Beijing, China. Chin. J. Osteoporosis 9, 134–136 (2003).
Wu, X. Y., Wu, X. P., Zhang, H., Cao, X. Z., Shan, P. F. & Liao, E. Y. Differences in male and female children and adolescents of acquisition with bone density in Changsha. Chin. J. Osteoporos 14, 865–874 (2008).
Rauchenzauner, M., Schmid, A., Heinz-Erian, P., Kapelari, K., Falkensammer, G., Griesmacher, A. et al. Sex- and age-specific reference curves for serum markers of bone turnover in healthy children from 2 months to 18 years. J. Clin. Endocrinol. Metab. 92, 443–449 (2007).
Sule, G., Campeau, P. M., Zhang, V. W., Nagamani, S. C., Dawson, B. C., Grover, M. et al. Next-generation sequencing for disorders of low and high bone mineral density. Osteoporos. Int. 24, 2253–2259 (2013).
Rauch, F., Lalic, L., Glorieux, F. H., Moffatt, P. & Roughley, P. Targeted sequencing of a pediatric metabolic bone gene panel using a desktop semiconductor next-generation sequencer. Calcif. Tissue Int. 95, 323–331 (2014).
Umair, M., Hassan, A., Jan, A., Ahmad, F., Imran, M., Samman, M. I. et al. Homozygous sequence variants in the FKBP10 gene underlie osteogenesis imperfecta in consanguineous families. J. Hum. Genet. 61, 207–213 (2016).
Murphy, L. A., Ramirez, E. A., Trinh, V. T., Herman, A. M., Anderson, V. C. & Brewster, J. L. Endoplasmic reticulum stress or mutation of an EF-hand Ca(2+)-binding domain directs the FKBP65 rotamase to an ERAD-based proteolysis. Cell Stress Chaperones 16, 607–619 (2011).
Lietman, C. D., Rajagopal, A., Homan, E. P., Munivez, E., Jiang, M. M., Bertin, T. K. et al. Connective tissue alterations in Fkbp10-/- mice. Hum. Mol. Genet. 23, 4822–4831 (2014).
Venturi, G., Monti, E., Dalle, C. L., Corradi, M., Gandini, A., Valenti, M. T. et al. A novel splicing mutation in FKBP10 causing osteogenesis imperfecta with a possible mineralization defect. Bone 50, 343–349 (2012).
Hug, N., Longman, D. & Caceres, J. F. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 44, 1483–1495 (2016).
Caparros-Martin, J. A., Valencia, M., Pulido, V., Martinez-Glez, V., Rueda-Arenas, I., Amr, K. et al. Clinical and molecular analysis in families with autosomal recessive osteogenesis imperfecta identifies mutations in five genes and suggests genotype-phenotype correlations. Am. J. Med. Genet. A 161A, 1354–1369 (2013).
Palomo, T., Fassier, F., Ouellet, J., Sato, A., Montpetit, K., Glorieux, F. H. et al. Intravenous bisphosphonate therapy of young children with osteogenesis imperfecta: skeletal findings during follow up throughout the growing years. J. Bone Miner. Res. 30, 2150–2157 (2015).
Otaify, G. A., Aglan, M. S., Ibrahim, M. M., Elnashar, M., El Banna, R. A. & Temtamy, S. A. Zoledronic acid in children with osteogenesis imperfecta and Bruck syndrome: a 2-year prospective observational study. Osteoporos Int. 27, 81–92 (2016).
Evelise, B. & Jay, R. S. Bisphosphonate treatment of children and adults with osteogenesis imperfecta: unanswered questions. Calcif. Tissue Int. 97, 101–103 (2015).
Acknowledgements
This study is supported by grants from the National Natural Science Foundation of China (81100623 and 81570802) and National Key Program of Clinical Science (WBYZ2011-873). We appreciate our patients and their families for their invaluable cooperation and participation in this research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Xu, Xj., Lv, F., Liu, Y. et al. Novel mutations in FKBP10 in Chinese patients with osteogenesis imperfecta and their treatment with zoledronic acid. J Hum Genet 62, 205–211 (2017). https://doi.org/10.1038/jhg.2016.109
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2016.109
This article is cited by
-
FKBP10 promotes proliferation of glioma cells via activating AKT-CREB-PCNA axis
Journal of Biomedical Science (2021)
-
Long-Term Follow-Up Outcomes of 19 Patients with Osteogenesis Imperfecta Type XI and Bruck Syndrome Type I Caused by FKBP10 Variants
Calcified Tissue International (2021)
-
Health-related quality of life in children with osteogenesis imperfecta: a large-sample study
Osteoporosis International (2019)
-
Novel Mutations in PLOD2 Cause Rare Bruck Syndrome
Calcified Tissue International (2018)
-
Novel compound heterozygous mutations in SERPINH1 cause rare autosomal recessive osteogenesis imperfecta type X
Osteoporosis International (2018)
