
Abstract
Oculocutaneous albinism (OCA) is characterized by hypopigmentation of the skin, hair and eye, and by ophthalmologic abnormalities caused by a deficiency in melanin biosynthesis. OCA type IV (OCA4) is one of the four commonly recognized forms of albinism, and is determined by mutation in the SLC45A2 gene. Here, we investigated the genetic basis of OCA4 in an Italian child. The mutational screening of the SLC45A2 gene identified two novel potentially pathogenic splicing mutations: a synonymous transition (c.888G>A) involving the last nucleotide of exon 3 and a single-nucleotide insertion (c.1156+2dupT) within the consensus sequence of the donor splice site of intron 5. As computer-assisted analysis for mutant splice-site prediction was not conclusive, we investigated the effects on pre-mRNA splicing of these two variants by using an in vitro minigene approach. Production of mutant transcripts in HeLa cells demonstrated that both mutations cause the almost complete abolishment of the physiologic donor splice site, with the concomitant unmasking of cryptic donor splice sites. To our knowledge, this work represents the first in-depth molecular characterization of splicing defects in a OCA4 patient.
Similar content being viewed by others


Introduction
Albinism is a heterogeneous group of inherited genetic diseases, affecting all ethnic backgrounds with an overall prevalence of approximately 1/17 000 people.1 Oculocutaneous albinism (OCA) is characterized by a general hypopigmentation of the skin, hair and eye, and by ophthalmologic abnormalities caused by a deficiency in melanin biosynthesis. Eye and optic system abnormalities include various degrees of congenital nystagmus, hypopigmentation of iris leading to iris translucency, reduced pigmentation of the retinal pigment epithelium, foveal hypoplasia, reduced visual acuity and refractive errors. Photophobia may be prominent. A characteristic feature is the misrouting of the optic nerves, consisting in an excessive crossing of the fibres in the optic chiasma, which can result in strabismus and reduced stereoscopic vision.2 These clinical signs are common to all types of albinism and are probably related to melanin reduction during embryonic development and early postnatal life.3
OCA is an autosomal recessive inherited condition; the four principal OCA forms (OCA1, OCA2, OCA3 and OCA4) are diagnosed based on the presence of mutations in the TYR, OCA2, TYRP1 and SLC45A2 genes, respectively.4, 5, 6, 7, 8 Recently, three additional loci involved in albinism (OCA5–7) have been identified. In particular, a novel OCA locus on chromosome 4q24 was found by linkage analysis in a large consanguineous Pakistani pedigree, but the causal gene (termed OCA5) has not been identified yet.9 Conversely, two genes involved in melanosome maturation and melanocyte differentiation, namely SLC24A5 and C10orf11,10, 11, 12 were identified as responsible for OCA6 and OCA7.13 However, few data about these recently described OCA forms are currently available. In addition, an X-linked form of ocular albinism (OA1), in which the phenotype is mainly restricted to eyes and optic system, is associated with mutations in the GPR143 gene.14
OCA4 (OMIM *606202, #606574) is worldwide distributed with a prevalence of 1:100 000 in most populations,7, 15, 16, 17, 18, 19 but in Japan, it is the most common form, accounting for about 24% of OCA cases.20 The causative gene is SLC45A2 (solute carrier family 45, member 2; also known as MATP gene), located on chromosome 5p13.3 and coding for a membrane-associated transporter protein.7 MATP is thought to be important for the proper processing and trafficking of melanosomal proteins Tyr, Tyrp1, Dct (Tyrp2), and seems to act as a proton-dependent transporter at the melanosomal membrane, showing a possible relationship with the P protein (OCA2).21, 22 Moreover, recent data based on experiments of heterologous expression of MATP in Saccharomyces cerevisiae suggested that it also acts as a sucrose transporter.23
OCA4 clinical phenotype is characterized by a high degree of heterogeneity, ranging from profound hypopigmentation (similar to OCA1A) to near normal skin colour. Indeed, some individuals accumulate pigmentation with age, whereas others remain completely hypopigmented throughout their life.13, 20, 24 To date, more than 80 mutations have been identified in the SLC45A2 gene.25
In this study, we report the identification of two hitherto-unknown splice defects underlying OCA4 in one Italian proband. The functional consequences of both mutations were elucidated by in vitro expression experiments.
Patients and Methods
Patient
The patient was initially recruited by the Genetic Unit and Ophthalmology Center of A.O.U. Meyer of Florence, Italy. Subsequently, the patient with his parents, came to Niguarda Ca’ Granda Hospital to undergo a multidisciplinary diagnostic workup including ophthalmological, dermatological, ENT (ear, nose and throat) evaluation and genetic counselling.
This study was conducted according to the Declaration of Helsinki and to the Italian legislation on sensible data recording. A signed informed consent for the genetic analysis was obtained from the proband’s parents.
Ophthalmologic evaluation
The patient underwent full ophthalmological and instrumental evaluations, including motor and sensory status, visual acuity, cycloplegic refraction, iris transillumination with slitlamp biomicroscope and macular translucency with indirect ophthalmoscope. The visual acuity assessment for distance and near was evaluated in monocular and binocular vision with appropriate tests (Pigassou figures, E test and Snellen charts).
The following instrumental examinations were performed: optical coherence tomography, visual evoked potential and electroretinogram. Spectral Domain OCT (Heildelberg, Germany) was used to evaluate the macular morphological characteristics, with particular reference to the presence/absence of foveal depression. Electrophysiological tests (Retimax, CSO, Scandicci, Italy) were performed to evaluate the optic nerve fiber decussation.
Genetic analysis
DNA was extracted from peripheral blood according to standard protocols.
The entire coding region, as well as the adjacent intronic sequences of TYR (NM_000372.4), OCA2 (NM_000275.2) and SLC45A2 (NM_016180.3) genes were PCR amplified from genomic DNA following standard amplification protocols. Amplified products were sequenced on both strands (primer sequences and experimental conditions available on request). Sequencing analysis was performed on an ABI Prism 3730 genetic analyzer (Life Technologies Corporation, Carlsbad, CA, USA) and the SeqScape software (Life Technologies Corporation) was used for mutation detection.
To screen for the presence of TYR and OCA2 exons rearrangements (deletion/duplication), the MLPA (Multiplex Ligation-dependent Probe Amplification) kit SALSA P325 (MRC-Holland, Amsterdam, the Netherlands) was used following the manufacturer’s instructions. The individual peak corresponding to each exon was identified based on the difference in migration relative to the size standards 500 LIZ™ (Life Technologies Corporation). The peak area of each fragment was compared with that of three control samples. Raw data were analyzed using the Coffalyser software v.9 (MRC-Holland, Amsterdam, the Netherlands). Each positive result was confirmed by two independent experiments and on two different DNA extractions.
In vitro analysis of splicing mutations
Computer-assisted analysis for splice-site prediction was accomplished using the NNSPLICE 0.9 (http://www.fruitfly.org/seq_tools/splice.html) and the Human Splicing Finder (HSF, http://www.umd.be/HSF/) programs.
For the functional characterization of the two newly identified mutations in SLC45A2, the relevant genomic DNA region was cloned in the hybrid alpha-globin-fibronectin minigene plasmid (pBS-KS_modified), in which the alternatively spliced extra-domain-B exon of fibronectin was removed to generate a site for the insertion of the exon under study.26 In particular, for the analysis of the c.888G>A substitution, a 746-bp fragment of SLC45A2 (introns 2 to 4) was PCR amplified from the patient’s genomic DNA using the following primers: OCA4_ex3_NdeI_F 5′-ggaattccatatgAGCCCAAGATCACAGCAAGT-3′ and OCA4_ex3_NdeI_R 5′-ggaattccatatgGGACCAGAGGAAAATGCAGA-3′ and cloned into the pBS-KS_modified vector (lowercase letters indicate nucleotides added to the primers to introduce the NdeI restriction site).
Concerning the c.1156+2dupT variant, a 572-bp region of SLC45A2 (including exon 5 with its flanking intronic sequences) was PCR amplified using the following primer pair OCA4_ex5_NdeI_F 5′-ggaattccatatgGAGATCTGATGCAGCAAGCA-3′ and OCA4_ex5_NdeI_R 5′-ggaattccatatgGGAACCCACTGATTCCAAGA-3′, and cloned into the pBS-KS_modified vector.
The obtained wild-type (pBS-KS-OCA4_ex3_wt and pBS-KS-OCA4_ex5_wt) and mutant (pBS-KS-OCA4_ex3_mut and pBS-KS-OCA4_ex5_mut) plasmids were isolated by the PureYield Plasmid Miniprep System (Promega, Madison, WI, USA). The correct orientation of the two inserts, as well as the presence/absence of either the c.888G>A or the c.1156+2dupT variant, were verified by direct DNA sequencing.
Recombinant plasmids were used for splicing assays, by transiently transfecting 4 μg of each minigene construct in HeLa cells (cultured according standard procedures). In each experiment, an equal number of cells (2.5 × 105) were transfected with the jetPRIME reagent (Euroclone, Wetherby, UK) in 6-well plates, as described by the manufacturer. Total RNA was isolated from the cells 24 h after transfection, using the Eurozol kit (Euroclone). Random hexamers and the ImProm-II Reverse Transcriptase (Promega) were used to perform first-strand cDNA synthesis starting from 500 ng of total RNA, according to the manufacturer's instructions. Of a total of 20 μl of the reverse-transcription (RT) reaction, 1 μl was used as template for amplifications, using primers annealing to the flanking α-globin/FN1 exonic sequences (α2-3 and Bra2 primers, see Figure 1a). RT-PCRs were performed under standard conditions using the GoTaq DNA Polymerase (Promega) on a Mastercycler EPgradient (Eppendorf, Hamburg, Germany).

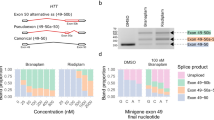
In vitro analysis of the effect of the c.888G>A (p.Q296Q) mutation on SLC45A2 pre-mRNA splicing. (a) Schematic representation of the hybrid pBS-KS-OCA4_ex3 minigene: α-globin and fibronectin (FN1) exons are represented by gray boxes, whereas introns are represented by lines (not to scale). The SLC45A2 exon 3 cloned, with its flanking intronic regions, into the NdeI site of the pBS-KS_modified vector is reported as a black box. The c.888G>A mutation, affecting the last nucleotide of exon 3, is indicated by a star. Primers used in RT-PCR experiments are indicated by arrows in correspondence of their position below the scheme of the minigene construct. (b) Left panel: RT-PCR products, obtained from RNA of HeLa cells transfected with the wild-type (wt) or the mutant (mut) minigene construct, separated on a 2% agarose gel, are shown. M: molecular weight marker (pUC9-HaeIII). Middle panel: the GeneMapper window shows fluorescence peaks corresponding to the molecular species amplified by RT-PCR. Red peaks represent the size standard (ROX-500 HD); blue peaks (shaded in gray) correspond to the RT-PCR-labeled products, whose abundance is reported on the right of the panel (%). The x axis indicates fluorescence units. Right panel: schematic representation of the obtained RT-PCR products (verified by Sanger sequencing). The length of each fragment is indicated.
Quantitation of splicing isoforms by fluorescent RT-PCR
To obtain a relative quantitation of the different isoforms generated by minigene assays, competitive fluorescent RT-PCRs were performed on RNA from transfected cells. To this aim, the same oligonucleotide pair adopted for splicing assays was used, with the reverse Bra2 primer labelled with the 6-FAM fluorophore. Amplified fragments were separated by capillary electrophoresis on an ABI-3130XL Genetic Analyzer (Life Technologies Corporation) and quantitated by the GeneMapper v4.0 software (Life Technologies Corporation). The sum of all fluorescence peak areas in a single run was set equal to 100%, and the relative quantity of each transcript expressed as a fraction of the total.
Results
Clinical features of the patient
The proband is a 3-year-old male of Italian origin. He has blonde platinum hair, pale skin and pale grey-blue iris. At 13 months, he had normal pupillary responses, horizontal nystagmus, astigmatism, no strabismus, iris transillumination (II-III grade of the Summers scale), foveal hypoplasia and albinotic fundus. Visual acuity could not be determined for low collaboration. No other apparent malformation was found at physical examination.
No consanguineous marriage was claimed by the parents.
The mutational screening of the proband revealed two novel putative splicing mutations in SLC45A2
The molecular screening of the patient was performed first by analyzing TYR and OCA2 genes, which represent the most frequently mutated OCA genes in the Italian population.27 Neither sequencing nor MLPA analysis of both genes revealed any point mutation or exon rearrangement. Subsequently, we analyzed the SLC45A2 gene and identified two novel putative pathogenic variants. The first one, a c.888G>A transition, is a synonymous change (p.Q296Q) that involves the last nucleotide of exon 3, suggesting that it might affect splicing. The second one, a c.1156+2dupT, is a single-nucleotide insertion within the consensus sequence of the donor splice site of intron 5 (IVS5+2dupT). The proband’s father was heterozygous for the c.888G>A variant, while the mother was heterozygous for the c.1156+2dupT insertion.
These two newly identified variants were absent in the cohort of 6503 European American and African American individuals, whose exome data are freely available through the Exome Variant Server.28
In vitro characterization of the two novel variants demonstrate their effect on splicing
The in silico analysis for the c.888G>A transition was performed by two different computer-assisted software for splice-site prediction: HSF detected the wild-type donor splice-site of exon 3 with a score of 85.90 and the mutated one with a score of 75.32. The NNSPLICE 0.9 software detected the wild-type donor splice-site with a score of 0.94, whereas, in the mutant sequence, it was no longer identified.
Given the unavailability of a suitable biological specimen to extract RNA, exon 3 of the SLC45A2 gene with the surrounding intronic sequences was cloned, either in the wild-type or in the mutant version, into a pBS-KS_modified hybrid minigene vector (Figure 1a). The obtained constructs (pBS-KS-OCA4_ex3_wt and pBS-KS-OCA4_ex3_mut) were transiently transfected into HeLa cells and SLC45A2 splicing products were analyzed by RT-PCR. All amplified fragments were then subjected to direct sequencing to characterize the aberrant splicing events. Indeed, transfection with the mutant vector gave rise to multiple aberrant products (Figure 1b). One PCR product was larger than the expected wild-type one (1222 vs 569 bp). In addition, four amplicons shorter than the wild type (329, 315, 243, and 179 bp long) were detected: (i) the first two caused by the retention of a shorter exon 3 because of the activation of two cryptic exonic donor splice sites, 86 and 72 bp downstream of the wild-type splice acceptor site, respectively; (ii) the third corresponded to the complete skipping of exon 3; and (iii) the last one resulted from the skipping of a part of fibronectin exon 24 together with the entire SLC45A2 exon 3, due to the activation of an exonic cryptic donor splice site (Figure 1b). While the skipping of the entire exon 3 or the inclusion of a shorter 72-bp-long exon 3 are predicted to cause a frameshift (leading to the introduction of a premature stop codon after 56 or 80 amino acids, respectively), the insertion of a 86-bp-long exon 3 is expected to determine the in-frame deletion of 80 amino acids. The relative quantitation by competitive fluorescent RT-PCR of all splicing isoforms generated by the minigene assay evidenced the presence of a residual amount (about 3%) of the wild-type transcript (Figure 1b).
The effect of the putative splicing mutation c.1156+2dupT was analyzed with the same approach. Computer-assisted splice-site analysis using HSF predicted a score reduction of the donor splice site (from 84.38 to 65.26) in the presence of the duplication. Moreover, a stronger cryptic donor splice site (located 72 bp downstream of the physiologic acceptor splice site of exon 5) was predicted. Instead, NNSPLICE 0.9 predicted the complete abolishment of the mutant donor splice-site compared with the wild type (whose score was 0.99).
Subsequently, appropriate minigene constructs were generated by cloning the relevant PCR-amplified genomic fragment (from intron 4 to 5) either in the wild-type or in the mutant version. Transfection with the mutant vector gave rise to a single splicing product, shorter than the expected wild-type one (315 vs 367 bp). Direct DNA sequencing confirmed that this aberrant product is due to the activation of the exonic cryptic donor splice site identified by the HSF in silico analysis. This aberrant splicing event would result in a frameshift followed by the introduction of a premature stop at codon 380 of the mutant protein, preceded by an abnormal sequence of 11 amino acids (p.V369Tfs*12) (Figure 2).

In vitro analysis of the effect of the c.1156+2dupT mutation on SLC45A2 pre-mRNA splicing. (a) Schematic representation of the hybrid pBS-KS-OCA4_ex5 minigene: α-globin and fibronectin (FN1) exons are represented by gray boxes, whereas introns are represented by lines (not to scale). The SLC45A2 exon 5 cloned, with its flanking intronic regions, into the NdeI site of the pBS-KS_modified vector is reported as a black box. The c.1156+2dupT insertion, affecting the donor splice site of exon 5, is indicated by a star. Primers used in RT-PCR experiments are indicated by arrows in correspondence of their position below the scheme of the minigene construct. (b) Left panel: RT-PCR products, obtained from RNA of HeLa cells transfected with the wild-type (wt) or the mutant (mut) minigene construct, separated on a 2% agarose gel, are shown. M: molecular weight marker (pUC9-HaeIII). Right panel: schematic representation of the obtained RT-PCR products (verified by Sanger sequencing). The length of each fragment is indicated.
Discussion
In this study, we analyzed the genetic defects underlying OCA4 in an Italian albino patient in whom we identified two novel splicing mutations, one of which is a synonymous transition (c.888G>A, p.Q269Q) involving the last nucleotide of exon 3 while the second one (c.1156+2dupT) lies in the consensus sequence of the donor splice site of SLC45A2 intron 5 (IVS5+2dupT).
We applied both in silico prediction and in vitro functional validation to experimentally verify the possible effect of the newly identified mutations on SLC45A2 pre-mRNA splicing. For both mutations, computer-assisted splice-site prediction analysis recognized a significant difference between the wild-type and the mutant sequences, suggesting that they might impact SLC45A2 splicing. These predictions were tested by a well-validated in vitro hybrid minigene approach,26, 29, 30 which we have recently applied to the study of two novel splicing mutations in the OCA2 gene.31
Functional analysis demonstrated that both mutations inactivate the relevant physiologic donor splice site and, at the same time, may lead to the activation of cryptic sites. In particular, the c.888G>A mutation seems to induce multiple aberrant splicing events, ranging from intron retention, complete exon skipping and activation of cryptic donor splice sites. Noteworthy, this is the second exonic splicing mutation identified and characterized in a OCA gene. Instead, the main functional consequence of the c.1156+2dupT mutation on SLC45A2 splicing is the unmasking of a cryptic exonic donor splice site, leading to the inclusion of a shorter out-of-frame exon 5. Although the in vitro characterization of splicing mutations might not completely mirror/recapitulate the actual splicing events occurring in vivo, it certainly provides a strong support to the pathogenic role of the identified variants. Indeed, minigene experiments have clearly evidenced a nearly complete loss of the wild-type transcript determined by the presence of either mutation (Figures 1 and 2). At the same time, most of the aberrant isoforms that might be produced in vivo as a consequence of the two identified mutations are predicted to cause the introduction of a premature termination codon, which might cause the transcript to be recognized and degraded by the nonsense-mediated mRNA decay mechanism.32
The activation of multiple cryptic splice sites within exon 3 observed in minigene studies prompted us to investigate the recognition of this exon in physiologic conditions. Interestingly, three SLC45A2 mRNA isoforms skipping exon 3 are annotated in GenBank (accession numbers: BC003597 deriving from melanoma; CU678525 and CU678524 deriving from the ORFeome project) (Supplementary Figure 1), suggesting that these splicing events could also occur in vivo. In addition, in silico prediction with the HSF algorithm identifies a cryptic donor splice site within exon 3 having a score (80.73) comparable to that of the physiologic one (85.9). The use of this cryptic splice site would lead to the inclusion of a 86-bp-long in-frame exon 3, corresponding to one of the aberrant isoforms detected in our minigene experiments (Figure 1b and Supplementary Figure 1), again suggesting its possible occurrence in vivo in physiologic conditions. Therefore, we attempted to characterize the splicing events involving exon 3 by RT-PCR assays performed on RNA extracted from hair follicles of a non-albino control. These experiments allowed the identification, beside the wild-type transcript, of an additional SLC45A2 isoform. Sequencing revealed that such aberrant product corresponds to the inclusion of the 86-bp-long in-frame exon 3 (Supplementary Figure 1), confirming that the predicted cryptic donor splice site can be recognized even in the absence of the c.888G>A mutation. We cannot exclude that the skipping of the whole exon 3 could also occur physiologically, although its detection can be hampered by the concomitant effect of nonsense-mediated mRNA decay on this out-of-frame isoform. Indeed, evidence supporting nonsense-mediated mRNA decay as a mechanism controlling SLC45A2 mRNA levels comes also from studies on the orthologous gene in birds.33 Taken together, these observations seem to suggest that exon 3 might be ‘leaky’ in recognition during mRNA splicing process.
References
Grønskov, K., Ek, J. & Brondum-Nielsen, K. Oculocutaneous albinism. Orphanet. J. Rare Dis. 2, 43–50 (2007).
Kirkwood, B. J. Albinism and implications with vision. Insight 34, 13–16 (2009).
King, R. A., Hearing, V. J., Creel, D. J. & Oetting, W. S. Albinism. Scriver, C. R. et al. The Metabolic and Molecular Basis of Inherited Disease, 8th edn. Vol.II, 5587–5627 (McGraw-Hill, New York, NY, USA, 2001).
Tomita, Y., Takeda, A., Okinaga, S., Tagami, H. & Shibahara, S. Human oculocutaneous albinism caused by single base insertion in the tyrosinase gene. Biochem. Biophys. Res. Commun. 164, 990–996 (1989).
Rinchik, E. M., Bultman, S. J., Horsthemke, B., Lee, S. T., Strunk, K. M., Spritz, R. A. et al. A gene for the mouse pink-eyed dilution locus and for human type II oculocutaneous albinism. Nature 361, 72–76 (1993).
Boissy, R. E., Zhao, H., Oetting, W. S., Austin, L. M., Wildenberg, S. C., Boissy, Y. L. et al. Mutation in and lack of expression of tyrosinase-related protein-1 (TRP-1) in melanocytes from an individual with brown oculocutaneous albinism: a new subtype of albinism classified as "OCA3”. Am. J. Hum. Genet. 58, 1145–1156 (1996).
Newton, J. M., Cohen-Barak, O., Hagiwara, N., Gardner, J. M., Davisson, M. T., King, R. A. et al. Mutations in the human orthologue of the mouse underwithe gene (uw) underlie a new form of Oculocutaneous albinism, OCA4. Am. J. Hum. Genet. 69, 981–988 (2001).
Gargiulo, A., Testa, F., Rossi, S., Di Iorio, V., Fecarotta, S., de Berardinis, T. et al. Molecular and clinical characterization of albinism in a large cohort of Italian patients. Invest. Ophthalmol. Vis. Sci. 52, 1281–1289 (2011).
Kausar, T., Bhatti, M. A., Ali, M., Shaikh, R. S. & Ahmed, Z. M. OCA5, a novel locus for non-syndromic oculocutaneous albinism, maps to chromosome 4q24. Clin. Genet. 84, 91–93 (2013).
Morice-Picard, F., Lasseaux, E., François, S., Simon, D., Rooryck, C., Bieth, E. et al. SLC24A5 mutations are associated with non-syndromic oculocutaneous albinism. J. Invest. Dermatol. 134, 568–571 (2014).
Wei, A. H., Zang, D. J., Zhang, Z., Liu, X. Z., He, X., Yang, L. et al. Exome sequencing identifies SLC24A5 as a candidate gene for nonsyndromic oculocutaneous albinism. J. Invest. Dermatol. 133, 1834–1840 (2013).
Grønskov, K., Dooley, C. M., Ostergaard, E., Kelsh, R. N., Hansen, L., Levesque, M. P. et al. Mutations in c10orf11, a melanocyte-differentiation gene, cause autosomal-recessive albinism. Am. J. Hum. Genet. 92, 415–421 (2013).
Montoliu, L., Grønskov, K., Wei, A. H., Martínez-García, M., Fernández, A., Arveiler, B. et al. Increasing the complexity: new genes and new types of albinism. Pigment Cell Melanoma Res. 27, 11–18 (2014).
Schiaffino, M. V., Bassi, M. T., Galli, L., Renieri, A., Bruttini, M., De Nigris, F. et al. Analysis of the OA1 gene reveals mutations in only one-third of patients with X-linked ocular albinism. Hum. Mol. Genet. 4, 2319–2325 (1995).
Rundshagen, U., Zuhlke, C., Opitz, S., Schwinger, E. & Käsmann-Kellner, B. Mutations in the MATP gene in five German patients affected by oculocutaneous albinism type 4. Hum. Mutat. 23, 106–110 (2004).
Sengupta, M., Chaki, M., Arti, N. & Ray, K. SLC45A2 variations in Indian oculocutaneous albinism patients. Mol. Vis. 13, 1406–1411 (2007).
Suzuki, T., Inagaki, K., Fukai, K., Obana, A., Lee, S. T. & Tomita, Y. A Korean case of oculocutaneous albinism type IV caused by a D157N mutation in the MATP gene. Br. J. Dermatol. 152, 174–175 (2005).
Grønskov, K., Ek, J., Sand, A., Scheller, R., Bygum, A., Brixen, K. et al. Birth prevalence and mutation spectrum in Danish patients with autosomal recessive albinism. Invest. Ophthalmol. Vis. Sci. 50, 1058–1064 (2009).
Konno, T., Abe, Y., Kawaguchi, M., Storm, K., Biervliet, M., Courtens, W. et al. Oculocutaneous albinism type IV: a boy of Moroccan descent with a novel mutation in SLC45A2. Am. J. Med. Genet. 149A, 1773–1776 (2009).
Inagaki, K., Suzuki, T., Shimizu, H., Ishii, N., Umezawa, Y., Tada, J. et al. Oculocutaneous albinism type 4 is one of the most common types of albinism in Japan. Am. J. Hum. Genet. 74, 466–471 (2004).
Costin, G. E., Valencia, J. C., Vieira, W. D., Lamoreux, M. L. & Hearing, V. J. Tyrosinase processing and intracellular trafficking is disrupted in mouse primary melanocytes carrying the underwhite (uw) mutation. A model for oculocutaneous albinism (OCA) type 4. J. Cell Sci. 116, 3203–3212 (2003).
Cullinane, A. R., Vilboux, T., O'Brien, K., Curry, J. A., Maynard, D. M., Carlson-Donohoe, H. et al. Homozygosity mapping and whole-exome sequencing to detect SLC45A2 and G6PC3 mutations in a single patient with oculocutaneous albinism and neutropenia. J. Invest. Dermatol. 131, 2017–2025 (2011).
Bartölke, R., Heinisch, J. J., Wieczorek, H. & Vitavska, O. Proton-associated sucrose transport of mammalian Solute Carrier Family 45: an analysis in Saccharomyces cerevisiae. Biochem. J. 464, 193–201 (2014).
Suzuki, T. & Tomita, Y. Recent advances in genetic analyses of oculocutaneous albinism type 2 and 4. J. Dermatol. Sci. 51, 1–9 (2008).
HGMD Professional http://biobase-international.com/hgmd/pro/all.php.
Baralle, M., Baralle, D., De Conti, L., Mattocks, C., Whittaker, J., Knezevich, A. et al. Identification of a mutation that perturbs NF1 agene splicing using genomic DNA samples and a minigene assay. J. Med. Genet. 40, 220–222 (2003).
Mártinez-García, M. & Montoliu, L. Albinism in Europe. J. Dermatol. 40, 39–24 (2013).
EVS, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA (URL http://evs.gs.washington.edu/EVS/; accessed January 2015.
Costantino, L., Rusconi, D., Soldà, G., Seia, M., Paracchini, V., Porcaro, L. et al. Fine characterization of the recurrent c.1584+18672A>G deep-intronic mutation in the cystic fibrosis transmembrane conductance regulator gene. Am. J. Respir. Cell Mol. Biol. 48, 619–625 (2013).
Paraboschi, E. M., Rimoldi, V., Soldà, G., Tabaglio, T., Dall'Osso, C., Saba, E. et al. Functional variations modulating PRKCA expression and alternative splicing predispose to multiple sclerosis. Hum. Mol. Genet. 23, 6746–6761 (2014).
Rimoldi, V., Straniero, L., Asselta, R., Mauri, L., Manfredini, E., Penco, S. et al. Functional characterization of two novel splicing mutations in the OCA2 gene associated with oculocutaneous albinism type II. Gene 537, 79–84 (2014).
Popp, M. W. & Maquat, L. E. Organizing principles of mammalian nonsense-mediated mRNA decay. Annu. Rev. Genet. 47, 139–165 (2013).
Gunnarsson, U., Hellström, A. R., Tixier-Boichard, M., Minvielle, F., Bed'hom, B., Ito, S. et al. Mutations in SLC45A2 cause plumage color variation in chicken and Japanese quail. Genetics 175, 867–877 (2007).
Acknowledgements
We thank Dr Emanuele Buratti (International Centre for Genetic Engineering and Biotechnology, Trieste, Italy) for kindly providing the hybrid pBS-KS vector. Laura Locatelli is also acknowledged for her technical assistance and enthusiasm. We would also like to thank the NHLBI GO Exome Sequencing Project and its ongoing studies which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926) and the Heart GO Sequencing Project (HL-103010). We also thank ‘Fondazione Paolina Brugnatelli’ for its valuable support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest. The authors alone are responsible for the content and writing of the paper.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Straniero, L., Rimoldi, V., Soldà, G. et al. Two novel splicing mutations in the SLC45A2 gene cause Oculocutaneous Albinism Type IV by unmasking cryptic splice sites. J Hum Genet 60, 467–471 (2015). https://doi.org/10.1038/jhg.2015.56
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.56
This article is cited by
-
Clinical utility gene card for oculocutaneous (OCA) and ocular albinism (OA)—an update
European Journal of Human Genetics (2021)
-
Evident hypopigmentation without other ocular deficits in Dutch patients with oculocutaneous albinism type 4
Scientific Reports (2021)
-
Clinical evaluation and molecular screening of a large consecutive series of albino patients
Journal of Human Genetics (2017)

{kind=link}