
Abstract
Human leukocyte antigen (HLA) polymorphisms are the most important genetic risk factors for rheumatoid arthritis (RA), a chronic systemic inflammatory disease of unknown etiology. Certain HLA-DRB1 alleles, known as shared epitope (SE) alleles because they have the same amino-acid sequence at positions 70–74, are associated with susceptibility to RA. A gene dosage effect is present for RA-predisposing SE alleles, and protective alleles show epistasis. An important role of amino-acid polymorphisms at positions 11 and 13 of the HLA-DRβ chain was also reported recently. Rheumatoid factor and anticitrullinated peptide antibodies are present in many RA patients. Similar to extra-articular manifestations, the presence of these autoantibodies is also associated with certain DRB1 alleles. Different frequencies of RA risk alleles in different ethnicities explain the varying prevalence of RA in different populations and suggest genetic heterogeneity of RA with regard to phenotype and population subsets. Some drug-induced hypersensitivity reactions due to disease-modifying antirheumatic drugs are also associated with HLA alleles. Understanding the role of HLA as the most important genetic factor relevant to RA susceptibility may help in determining its pathogenesis and pave the way to personalized medicine.
Similar content being viewed by others
Introduction
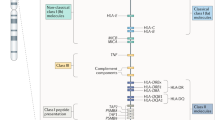
The human major histocompatibility complex is a region of ~3.6 Mbp on chromosome 6p21.31, comprising >200 genetic loci including the human leukocyte antigens (HLA).1 The main gene products of this region that are relevant to immunity are designated class I molecules (HLA-A, C and B) and class II molecules (HLA-DR, DQ and DP). The former consists of a type I transmembrane protein of 45 kD (the heavy chain) and a soluble protein of 12 kD (β2-microglobulin, the light chain). They bind and present intracellular antigens to the T-cell receptors of CD8+ T cells. In contrast, the latter consists of two type I transmembrane proteins (the 32 kD α chain and 28 kD β chain) and present both intra- and extracellular antigens to the T-cell receptors of CD4+ T cells. The HLA-A, C and B genes encode HLA-A, C and B heavy chains and the B2M gene, located elsewhere (on chromosome 15) encodes the β2-microglobulin light chain. The DRA1, DRB1 genes (and additionally DRB3, DRB4 and DRB5 genes in some haplotypes), and the DQA1, DQB1, DPA1 and DPB1 genes encode the α and β chains of HLA-DR, DQ and DP molecules, respectively. HLA is hyperpolymorphic and >12 000 distinct alleles of these genes have been identified (IMGT/HLA data base, http://www.ebi.ac.uk/ipd/imgt/hla/). Studies of the frequencies of HLA alleles have revealed genetic associations with >100 diseases. Most of the different genes of the major histocompatibility complex are in strong linkage disequilibrium, making it difficult to identify the primary causative gene.
Rheumatoid arthritis (RA) is a chronic systemic inflammatory disease of unknown etiology. It mainly affects synovial joints, reducing quality of life and life expectancy. The development of extra-articular manifestations, including serositis, Felty’s syndrome, rheumatoid vasculitis, lymphoproliferative disease and interstitial lung disease (ILD), is frequent complications. RA is thought to represent a set of clinical syndromes comprising different disease entities with specific features. The pathogenesis of RA is likewise considered to be multifactorial, with disease susceptibility associated with genetic, environmental and stochastic factors.2 The prevalence of RA is 0.5–1.0% in most populations. The concordance rate in monozygotic twins is 12–30% and the sibling recurrence rate for RA lies between 5 and 10%. The heritability of RA is estimated to be 65%.3, 4, 5 The association of RA with HLA was already reported ~40 years ago.6 To date, more than 100 risk loci for RA have been identified in large-scale studies including genome-wide association studies.7 All of the known genetic risk factors together were estimated to explain 16% of RA risk, 11% of which was due to HLA alone.4, 8 The remaining 49% of genetic predisposition to RA represents missing heritability. Thus, HLA is the most important known genetic risk factor for RA.
HLA and RA
Many studies have reported that polymorphisms at the HLA-DRB1 locus are associated with several different autoimmune diseases including RA. The following HLA-DRB1 alleles are associated with RA in most ethnic groups: DRB1*04:01, *04:04, *04:05, *01:01 and *10:01. In Europeans DRB1*04:01 and in Eastern Asians *04:05 are most strongly associated with RA. Some other DRB1 alleles are also weakly associated with RA susceptibility. Almost all of these share amino-acid sequences at positions 70–74 (QKRAA, RRRAA or QRRAA) in the third hypervariable region of the HLA-DRβ chain. These are designated shared epitope (SE) alleles.9 DRB1*09:01 is not a SE allele, possessing RRRAE at position 70–74, but is nonetheless also associated with RA in some populations, especially in East Asians.10, 11, 12, 13 A different classification criterion for DRB1 alleles has been established, also based on amino-acid residues at positions 70–74, as follows: S1 (XA/ERAA), S2 (XKRAA), S3P (Q/RRRAA), S3D (DRRAA) and X (without XXRAA).14, 15 As S2 and S3P confer higher susceptibility for RA, S1, S3D and X can be pooled as ‘L alleles’. These amino-acid residues form the peptide position 4 binding pocket of the DR molecule. Thus, polymorphisms of these amino-acid residues may change the repertoire of the presented peptides.16
DRB1 is considered to be the primary gene for RA susceptibility. DRB1 and DQB1 are in strong linkage disequilibrium. This makes it difficult to exclude the hypothesis that the DQB1 allele is primarily responsible for the association with RA.17 Certain combinations of DRB1 alleles confer higher risk, such as heterozygosity for *04:01/*04:04 or *04:05/*09:01.13, 18 Such a heterozygote disadvantage was also reported in type 1 diabetes and systemic lupus erythematosus.19, 20 These data cannot exclude the involvement of trans-complementing heterodimers formed by DQA1 and DQB1 genes.
A gene dosage effect has been noted for the associations of SE alleles with susceptibility to RA. Thus, having two SE alleles confers a higher risk for RA and for more severe bone destruction. Because the molecular mechanisms responsible for the gene dosage effect are not clear, a cusp hypothesis was developed to explain it, as follows. SE molecules act as allele-specific signal transduction ligands that interact with non-HLA cell surface receptors, including cell surface calreticulin, for production of nitric oxide and reactive oxygen species for cell signaling.21
DRB1*13:01 and *13:02 are negatively associated with RA in European and East Asian populations, respectively.22, 23 The resistant effect of these DRB1*13 alleles is dominant over the susceptible effect of SE alleles in DRB1 heterozygotes. The protective DRB1 alleles are thought to be recognized by autoreactive T-cell receptors with higher affinity than predisposing DRB1 alleles. As a result, negative selection and development of DR-driven autoreactive regulatory T cells are promoted.24 An effect of noninherited maternal antigen was also reported as protective against RA. Thus, the protective DRB1 allele that was not inherited from the mother decreases RA susceptibility risk in her child.25 This phenomenon could be explained by the microchimerism of maternal cells that enter the circulation of the child during gestation or birth.
Recent analyses of the association of polymorphisms of amino-acid residues with RA showed important influences of positions 11 and 13 of the HLA-DRβ chain, followed by positions 71 and 74.26 The amino-acid residues at positions 11 and 13 of HLA-DRβ form the binding pocket for peptide position 6 of DR molecules.16 It can be concluded that of the many hypotheses proposed to explain the association of HLA-DRB1 with RA; some seem tentative, others credible, but none unequivocal.
HLA and subsets of RA
Rheumatoid factor (RF) is composed of autoantibodies specific for denatured IgG-Fc domains and is present in ~80% of RA patients, having some roles in the formation of immune complexes or cryoglobulin, which cause some types of RA symptoms. RF has long been used as a biomarker for RA in daily clinical practice, but is often detected in aged individuals without RA. An association of SE with RF-positive RA has been established; however, only a weak association with RF-negative RA has been established.27 A recent study showed that DR molecules form complexes with intracellular misfolded proteins without processing and are transferred to the cell surface. The IgG heavy chain was similarly presented by DR molecules with different allele-specific binding affinity,28 explaining the mechanism by which RF is generated and the important role of SE alleles.
Anticitrullinated peptide antibodies (ACPA) are present in 70–80% of RA patients. Although RF and ACPA are frequently found in the same patient, the specificity of ACPA for diagnosing RA is higher than RF. The presence of ACPA is considered a predictive marker of bone destruction; however, it is still controversial as to whether ACPA can also predict the response to treatment with biologic or nonbiologic disease-modifying antirheumatic drugs.29, 30, 31, 32 Because of their high specificity, ACPA are believed to have some role in the pathogenesis of RA. SE alleles are associated with ACPA-positive RA, but only relatively weakly with ACPA-negative RA.4, 23, 27, 33, 34 Other than SE alleles, DRB1*03 and DRB1*14:54, are associated with ACPA-negative RA in European and Asian populations, respectively.23, 34, 35 The differential contribution of HLA was also confirmed in a genome-wide association study between ACPA-positive and -negative RA susceptibility.36, 37 An analysis of the association of polymorphisms of amino-acid residues with ACPA-positive and -negative RA also showed different specificities,38 suggesting the distinct disease category of ACPA-positive and -negative RA. Gene–environment interactions were detected between SE alleles and smoking for susceptibility to ACPA-positive RA.39, 40, 41 Citrullinated peptides were found in the lung of individuals with a smoking habit, and are believed to be generated by smoking. The citrullinated peptides are thought to be autoantigens presented by SE alleles in the pathogenesis of RA.42
Autoantibodies to Ro/SS-A and La/SS-B, components of ribonucleoprotein complexes, are specifically present in RA patients with secondary Sjögren’s syndrome that affects salivary and lacrimal glands, causing dry mouth or dry eye. Anti-Ro/SS-A antibodies are present in ~20% of RA patients and anti-La/SS-B antibodies in ~5%. Anti-Ro/SS-A antibodies are detected in almost all RA patients with anti-La/SS-B antibodies. The DRB1*08:03-DQB1*06:01-DPB1*05:01 haplotype is strongly associated with anti-Ro/SS-A antibodies in Japanese RA patients, whereas DRB1*15:01-DQB1*06:02-DPB1*05:01 is associated with anti-La/SS-B, suggesting the primary role of DPB1*05:01 for the production of autoantibodies to ribonucleoprotein.43
Extra-articular manifestations are frequent complications of RA and influence its prognosis. Associations of extra-articular manifestations with the SE or DRB1*04 alleles have been reported.44, 45, 46, 47 However, protective effects of SE and the predispositional effects of the DR2 serological group (DRB1*15 and *16) for ILD are seen in the Japanese RA population,48, 49, 50 although a weak protective effect of SE for ILD in RA has also been reported.48, 50, 51 Finally, DR4 is also associated with airway disease in RA.52
It was well known that the development of lymphoproliferative disorders is frequent in RA patients, although the incidence of some other malignancies in RA is relatively lower.53 A tendency toward a higher frequency of DQB1*06:01 was noted in RA patients with lymphoproliferative disorders (Table 1, Furukawa H et al, unpublished results), although the effect was not statistically significant. When the analysis was restricted to patients treated with methotrexate, the frequency of HLA-B*15:11 was significantly higher.54 Thus, genetic association studies of RA subsets with HLA support the notion that RA is a phenotypically heterogeneous syndrome.
The prevalence of RA is different in different ethnic populations, at ~1% in Europeans, ~0.5% in Asian or African populations, but >2% in some populations of Amerindians.55, 56 The main SE alleles are also differently associated with RA in the different populations. Thus, DRB1*04:01 and *04:05 are most strongly associated with RA in European and East Asian populations, respectively. In addition, the effect size of SE alleles on associations with RA is weaker in some populations.57, 58, 59 Because the frequency of DRB1*09:01 is low in European populations, an association of this allele with RA could not be readily observed. An association of DRB1*03 and DRB1*13:01 with ACPA-negative RA is seen in European populations,35, 60 whereas it is DRB1*04:05 and *14:54 that are associated with ACPA-negative RA in Japanese.23, 34 The differences in prevalence of RA among populations could be explained by the different frequencies of RA risk alleles, although the effects of environmental factors cannot be excluded.
Analyses of HLA associations with RA in Asian populations, mainly Chinese and Korean, revealed amino-acid residues, different from those of Europeans, conferring disease susceptibility.61 The involvement of amino-acid position 57 is noted in Chinese and Korean RA, instead of position 71. Similar amino-acid residues are associated with RA in the Japanese population (Oka et al., unpublished results). The genetic risk of possessing DRB1 in African Americans is explained by amino-acid positions 11 and 57 of the DRβ chain.62 The different frequencies of RA risk alleles in each population reflect these different amino-acid residues conferring risk. These data indicate that distinct disease entities observed in each ethnic population reflect the genetic heterogeneity of RA.
HLA and drug-induced hypersensitivity reactions in RA
A strong association between certain HLA alleles and the occurrence of adverse cutaneous reactions has been reported, including Stevens–Johnson syndrome and toxic epidermal necrolysis. These patients suffer from blisters and erosions on the skin and mucous membranes; distinguishing between the two syndromes is based on the affected body surface area. Stevens–Johnson syndrome and toxic epidermal necrolysis are known to be induced by several specific drugs, with striking associations with HLA alleles. In this regard, allopurinol effects are associated with B*58:01,63 abacavir with B*57:01,64, 65 carbamazepine with B*15:02 in Chinese but with A*31:01 in Japanese and Europeans66, 67, 68 and methazolamide with B*59:01 in Japanese.69 Associations with HLA alleles were also confirmed in other drug-induced hypersensitivity reactions. Agranulocytosis induced by methimazole is associated with DRB1*08:03.70 Cholestatic hepatotoxicity induced by ticlopidine and tiopronin is associated with A*33:03.71, 72 Liver injury induced by flucloxacillin is associated with B*57:01, and by amoxicillin–clavulanate with DRB1*15:01.73, 74 Myopathy induced by statins is associated with DRB1*11:01.75 Some of these associations are useful for predicting and preventing drug-induced hypersensitivity reactions by HLA typing before starting the medication. The mechanisms responsible for these drug hypersensitivity reactions so strongly associated with HLA remain unclear. Several possibilities can be considered. Acting as haptens, drugs or their metabolites may bind to peptides presented by HLA molecules and activate T-cell receptors. Alternatively, drugs may bridge between HLA molecules and T-cell receptors, independently of the peptide presented on HLA, and thus activate T cells.76 They may bind to the grooves of specific HLA alleles and alter the repertoire of presented peptides.77
Disease-modifying antirheumatic drugs have been used for the treatment of RA for many years. Some nonbiologic disease-modifying antirheumatic drugs often cause hypersensitivity reactions. For example, Stevens–Johnson syndrome, toxic epidermal necrolysis and drug-induced hypersensitivity syndrome are induced by sulfasalazine.78, 79, 80, 81 D-penicillamine, gold salts and bucillamine cause proteinuria due to membranous nephropathy.82, 83, 84, 85 Drug-induced ILD can be triggered by gold salts, methotrexate, leflunomide and tacrolimus.86, 87, 88, 89 The Japanese are believed to be more susceptible to drug-induced ILD than other ethnic groups, suggesting the involvement of genetic factors in its pathogenesis. Indeed, an association of HLA-A*31:01 with drug-induced ILD in methotrexate-treated Japanese RA patients has been reported.90 Proteinuria induced by D-penicillamine and gold salts is associated with DR3 in Europeans91 and bucillamine-induced proteinuria with DRB1*08:02 in Japanese.92 Further studies are likely to reveal associations with HLA alleles of other drug-induced hypersensitivity reactions caused by disease-modifying antirheumatic drugs.
Secondary Sjögren’s syndrome or the presence of anti-Ro/SS-A antibodies are thought to be predictive markers for the development of drug-induced hypersensitivity reactions in RA patients.93, 94 The association of DR3 with RA patients possessing anti-Ro/SS-A antibodies95 can explain why this specificity is associated with drug-induced proteinuria in Europeans.91, 96 However, an association of DRB1*08:03 with anti-Ro/SS-A antibodies43 cannot explain the correlation because it is the DRB1*08:02 allele that is associated with bucillamine-induced proteinuria in Japanese RA.92 Thus, genetic association studies on HLA could provide surrogate markers for drug-induced hypersensitivity reactions in some cases, but can also provide essential individual genetic markers useful for guiding RA treatment. These observations imply that HLA may have a substantial role in drug-induced hypersensitivity reactions in RA patients and provide important information for developing personalized medicine for RA.
Conclusion
Remarkable recent technical progress in genotyping methods has yielded major advances in our knowledge of the complex genetics of RA susceptibility. Despite the fact that HLA remains the most important genetic factor for the pathogenesis of RA, explanations for a large part of RA heritability are still lacking. Alternative assessments of HLA risk alleles may not be universally applicable to RA because of disease heterogeneity and ethnic diversity in its manifestations. These also make it difficult to predict the genetic risk for RA, its severity and response to treatment with disease-modifying antirheumatic drugs with sufficient accuracy. However, predictability of drug-induced hypersensitivity reactions is acceptably specific. To go beyond the status quo of disease association studies, the actual genetic and immunological mechanisms responsible for the heterogeneous manifestations of RA in the context of SE alleles still need to be clarified. To paraphrase Sir Arthur Conan Doyle, the words ‘of murder’ could be substituted by ‘between HLA and RA’ in the following sentence: ‘There's the scarlet thread of murder running through the colourless skein of life, and our duty is to unravel it, and isolate it, and expose every inch of it.’97
References
The MHC sequencing consortium Complete sequence and gene map of a human major histocompatibility complex. The MHC sequencing consortium. Nature 401, 921–923 (1999).
Perricone, C., Ceccarelli, F. & Valesini, G. An overview on the genetic of rheumatoid arthritis: a never-ending story. Autoimmun. Rev. 10, 599–608 (2011).
MacGregor, A. J., Snieder, H., Rigby, A. S., Koskenvuo, M., Kaprio, J. & Aho, K. et al. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum. 43, 30–37 (2000).
van der Woude, D., Houwing-Duistermaat, J. J., Toes, R. E., Huizinga, T. W., Thomson, W. & Worthington, J. et al. Quantitative heritability of anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis. Arthritis Rheum. 60, 916–923 (2009).
Scott, D. L., Wolfe, F. & Huizinga, T. W. Rheumatoid arthritis. Lancet 376, 1094–1108 (2010).
Stastny, P. Mixed lymphocyte cultures in rheumatoid arthritis. J. Clin. Invest. 57, 1148–1157 (1976).
Okada, Y., Wu, D., Trynka, G., Raj, T., Terao, C. & Ikari, K. et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506, 376–381 (2014).
Viatte, S., Plant, D. & Raychaudhuri, S. Genetics and epigenetics of rheumatoid arthritis. Nat. Rev. Rheumatol. 9, 141–153 (2013).
Gregersen, P. K., Silver, J. & Winchester, R. J. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 30, 1205–1213 (1987).
Gonzalez, A., Nicovani, S., Massardo, L., Bull, P., Rodriguez, L. & Jacobelli, S. Novel genetic markers of rheumatoid arthritis in Chilean patients, by DR serotyping and restriction fragment length polymorphism analysis. Arthritis Rheum. 35, 282–289 (1992).
Wakitani, S., Imoto, K., Murata, N., Toda, Y., Ogawa, R. & Ochi, T. The homozygote of HLA-DRB1*0901, not its heterozygote, is associated with rheumatoid arthritis in Japanese. Scand. J. Rheumatol. 27, 381–382 (1998).
Milicic, A., Lee, D., Brown, M. A., Darke, C. & Wordsworth, B. P. HLA-DR/DQ haplotype in rheumatoid arthritis: novel allelic associations in UK Caucasians. J. Rheumatol. 29, 1821–1826 (2002).
Lee, H. S., Lee, K. W., Song, G. G., Kim, H. A., Kim, S. Y. & Bae, S. C. Increased susceptibility to rheumatoid arthritis in Koreans heterozygous for HLA-DRB1*0405 and *0901. Arthritis Rheum. 50, 3468–3475 (2004).
du Montcel, S. T., Michou, L., Petit-Teixeira, E., Osorio, J., Lemaire, I. & Lasbleiz, S. et al. New classification of HLA-DRB1 alleles supports the shared epitope hypothesis of rheumatoid arthritis susceptibility. Arthritis Rheum. 52, 1063–1068 (2005).
Pratesi, F., Petit Teixeira, E., Sidney, J., Michou, L., Puxeddu, I. & Sette, A. et al. HLA shared epitope and ACPA: just a marker or an active player? Autoimmun. Rev. 12, 1182–1187 (2013).
Jardetzky, T. S., Brown, J. H., Gorga, J. C., Stern, L. J., Urban, R. G. & Chi, Y. I. et al. Three-dimensional structure of a human class II histocompatibility molecule complexed with superantigen. Nature 368, 711–718 (1994).
Singal, D. P., D'Souza, M., Reid, B., Bensen, W. G., Kassam, Y. B. & Adachi, J. D. HLA-DQ beta-chain polymorphism in HLA-DR4 haplotypes associated with rheumatoid arthritis. Lancet 2, 1118–1120 (1987).
Hall, F. C., Weeks, D. E., Camilleri, J. P., Williams, L. A., Amos, N. & Darke, C. et al. Influence of the HLA-DRB1 locus on susceptibility and severity in rheumatoid arthritis. Qjm 89, 821–829 (1996).
Erlich, H., Valdes, A. M., Noble, J., Carlson, J. A., Varney, M. & Concannon, P. et al. HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: analysis of the type 1 diabetes genetics consortium families. Diabetes 57, 1084–1092 (2008).
Shimane, K., Kochi, Y., Suzuki, A., Okada, Y., Ishii, T. & Horita, T. et al. An association analysis of HLA-DRB1 with systemic lupus erythematosus and rheumatoid arthritis in a Japanese population: effects of *09:01 allele on disease phenotypes. Rheumatology (Oxford) 52, 1172–1182 (2013).
Holoshitz, J. The quest for better understanding of HLA-disease association: scenes from a road less travelled by. Discov. Med. 16, 93–101 (2013).
van der Woude, D., Lie, B. A., Lundstrom, E., Balsa, A., Feitsma, A. L. & Houwing-Duistermaat, J. J. et al. Protection against anti-citrullinated protein antibody-positive rheumatoid arthritis is predominantly associated with HLA-DRB1*1301: a meta-analysis of HLA-DRB1 associations with anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis in four European populations. Arthritis Rheum. 62, 1236–1245 (2010).
Oka, S., Furukawa, H., Kawasaki, A., Shimada, K., Sugii, S. & Hashimoto, A. et al. Protective effect of the HLA-DRB1*13:02 allele in Japanese rheumatoid arthritis patients. PLoS ONE 9, e99453 (2014).
Tsai, S. & Santamaria, P. MHC class II polymorphisms, autoreactive T-cells, and autoimmunity. Front. Immunol. 4, 321 (2013).
Feitsma, A. L., Worthington, J., van der Helm-van Mil, A. H., Plant, D., Thomson, W. & Ursum, J. et al. Protective effect of noninherited maternal HLA-DR antigens on rheumatoid arthritis development. Proc. Natl Acad. Sci. USA 104, 19966–19970 (2007).
Raychaudhuri, S., Sandor, C., Stahl, E. A., Freudenberg, J., Lee, H. S. & Jia, X. et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat. Genet. 44, 291–296 (2012).
Morgan, A. W., Thomson, W., Martin, S. G., Carter, A. M., Erlich, H. A. & Barton, A. et al. Reevaluation of the interaction between HLA-DRB1 shared epitope alleles, PTPN22, and smoking in determining susceptibility to autoantibody-positive and autoantibody-negative rheumatoid arthritis in a large UK Caucasian population. Arthritis Rheum. 60, 2565–2576 (2009).
Jin, H., Arase, N., Hirayasu, K., Kohyama, M., Suenaga, T. & Saito, F. et al. Autoantibodies to IgG/HLA class II complexes are associated with rheumatoid arthritis susceptibility. Proc. Natl Acad. Sci. USA 111, 3787–3792 (2014).
Kroot, E. J., de Jong, B. A., van Leeuwen, M. A., Swinkels, H., van den Hoogen, F. H. & van't Hof, M. et al. The prognostic value of anti-cyclic citrullinated peptide antibody in patients with recent-onset rheumatoid arthritis. Arthritis Rheum. 43, 1831–1835 (2000).
Makinen, H., Hannonen, P. & Sokka, T. Sex: a major predictor of remission as measured by 28-joint Disease Activity Score (DAS28) in early rheumatoid arthritis? Ann. Rheum. Dis. 67, 1052–1053 (2008).
Klaasen, R., Cantaert, T., Wijbrandts, C. A., Teitsma, C., Gerlag, D. M. & Out, T. A. et al. The value of rheumatoid factor and anti-citrullinated protein antibodies as predictors of response to infliximab in rheumatoid arthritis: an exploratory study. Rheumatology (Oxford) 50, 1487–1493 (2011).
Mori, S., Hirose, J. & Yonemura, K. Contribution of HLA-DRB1*04 alleles and anti-cyclic citrullinated antibodies to development of resistance to disease-modifying antirheumatic drugs in early rheumatoid arthritis. Clin. Rheumatol. 29, 1357–1366 (2010).
Holoshitz, J. The rheumatoid arthritis HLA-DRB1 shared epitope. Curr. Opin. Rheumatol. 22, 293–298 (2010).
Terao, C., Ohmura, K., Ikari, K., Kochi, Y., Maruya, E. & Katayama, M. et al. ACPA-negative RA consists of two genetically distinct subsets based on RF positivity in Japanese. PLoS ONE 7, e40067 (2012).
Verpoort, K. N., van Gaalen, F. A., van der Helm-van Mil, A. H., Schreuder, G. M., Breedveld, F. C. & Huizinga, T. W. et al. Association of HLA-DR3 with anti-cyclic citrullinated peptide antibody-negative rheumatoid arthritis. Arthritis Rheum. 52, 3058–3062 (2005).
Padyukov, L., Seielstad, M., Ong, R. T., Ding, B., Ronnelid, J. & Seddighzadeh, M. et al. A genome-wide association study suggests contrasting associations in ACPA-positive versus ACPA-negative rheumatoid arthritis. Ann. Rheum. Dis. 70, 259–265 (2011).
Bossini-Castillo, L., de Kovel, C., Kallberg, H., van 't Slot, R., Italiaander, A. & Coenen, M. et al. A genome-wide association study of rheumatoid arthritis without antibodies against citrullinated peptides. Ann. Rheum. Dis. 74, e15 (2015).
Han, B., Diogo, D., Eyre, S., Kallberg, H., Zhernakova, A. & Bowes, J. et al. Fine mapping seronegative and seropositive rheumatoid arthritis to shared and distinct HLA alleles by adjusting for the effects of heterogeneity. Am. J. Hum. Genet. 94, 522–532 (2014).
Padyukov, L., Silva, C., Stolt, P., Alfredsson, L. & Klareskog, L. A gene-environment interaction between smoking and shared epitope genes in HLA-DR provides a high risk of seropositive rheumatoid arthritis. Arthritis Rheum. 50, 3085–3092 (2004).
Kallberg, H., Padyukov, L., Plenge, R. M., Ronnelid, J., Gregersen, P. K. & van der Helm-van Mil, A. H. et al. Gene-gene and gene-environment interactions involving HLA-DRB1, PTPN22, and smoking in two subsets of rheumatoid arthritis. Am. J. Hum. Genet. 80, 867–875 (2007).
Lundstrom, E., Kallberg, H., Alfredsson, L., Klareskog, L. & Padyukov, L. Gene-environment interaction between the DRB1 shared epitope and smoking in the risk of anti-citrullinated protein antibody-positive rheumatoid arthritis: all alleles are important. Arthritis Rheum. 60, 1597–1603 (2009).
Klareskog, L., Malmstrom, V., Lundberg, K., Padyukov, L. & Alfredsson, L. Smoking, citrullination and genetic variability in the immunopathogenesis of rheumatoid arthritis. Semin. Immunol. 23, 92–98 (2011).
Furukawa, H., Oka, S., Shimada, K., Sugii, S., Hashimoto, A. & Komiya, A. et al. Association of increased frequencies of HLA-DPB1*05:01 with the presence of anti-Ro/SS-A and anti-La/SS-B antibodies in Japanese rheumatoid arthritis and systemic lupus erythematosus patients. PLoS ONE 8, e53910 (2013).
Turesson, C., Weyand, C. M. & Matteson, E. L. Genetics of rheumatoid arthritis: is there a pattern predicting extraarticular manifestations? Arthritis Rheum. 51, 853–863 (2004).
Lanchbury, J. S., Jaeger, E. E., Sansom, D. M., Hall, M. A., Wordsworth, P. & Stedeford, J. et al. Strong primary selection for the Dw4 subtype of DR4 accounts for the HLA-DQw7 association with Felty's syndrome. Hum. Immunol. 32, 56–64 (1991).
Weyand, C. M., Xie, C. & Goronzy, J. J. Homozygosity for the HLA-DRB1 allele selects for extraarticular manifestations in rheumatoid arthritis. J. Clin. Invest. 89, 2033–2039 (1992).
Kim, H. Y., Min, J. K., Yang, H. I., Park, S. H., Hong, Y. S. & Jee, W. H. et al. The impact of HLA-DRB1*0405 on disease severity in Korean patients with seropositive rheumatoid arthritis. Br. J. Rheumatol. 36, 440–443 (1997).
Migita, K., Nakamura, T., Koga, T. & Eguchi, K. HLA-DRB1 alleles and rheumatoid arthritis-related pulmonary fibrosis. J. Rheumatol. 37, 205–207 (2010).
Furukawa, H., Oka, S., Shimada, K., Sugii, S., Ohashi, J. & Matsui, T. et al. Association of human leukocyte antigen with interstitial lung disease in rheumatoid arthritis: a protective role for shared epitope. PLoS ONE 7, e33133 (2012).
Mori, S., Koga, Y. & Sugimoto, M. Different risk factors between interstitial lung disease and airway disease in rheumatoid arthritis. Respir. Med. 106, 1591–1599 (2012).
Turesson, C., Schaid, D. J., Weyand, C. M., Jacobsson, L. T., Goronzy, J. J. & Petersson, I. F. et al. The impact of HLA-DRB1 genes on extra-articular disease manifestations in rheumatoid arthritis. Arthritis Res. Ther. 7, R1386–R1393 (2005).
Radoux, V., Menard, H. A., Begin, R., Decary, F. & Koopman, W. J. Airways disease in rheumatoid arthritis patients. One element of a general exocrine dysfunction. Arthritis Rheum. 30, 249–256 (1987).
Hashimoto, A., Chiba, N., Tsuno, H., Komiya, A., Furukawa, H. & Matsui, T. et al. Incidence of malignancy and the risk of lymphoma in japanese patients with rheumatoid arthritis compared to the general population. J. Rheumatol. (2015) 140533 (2015).
Yamakawa, N., Fujimoto, M., Kawabata, D., Terao, C., Nishikori, M. & Nakashima, R. et al. A clinical, pathological, and genetic characterization of methotrexate-associated lymphoproliferative disorders. J. Rheumatol. 41, 293–299 (2014).
Bridges, S. L. Jr ., Hughes, L. B., Mikuls, T. R., Howard, G., Tiwari, H. K. & Alarcon, G. S. et al. Early rheumatoid arthritis in African-Americans: the CLEAR Registry. Clin. Exp. Rheumatol. 21 (5 Suppl 31), S138–S145 (2003).
Alamanos, Y. & Drosos, A. A. Epidemiology of adult rheumatoid arthritis. Autoimmun. Rev. 4, 130–136 (2005).
Teller, K., Budhai, L., Zhang, M., Haramati, N., Keiser, H. D. & Davidson, A. HLA-DRB1 and DQB typing of Hispanic American patients with rheumatoid arthritis: the "shared epitope" hypothesis may not apply. J. Rheumatol. 23, 1363–1368 (1996).
McDaniel, D. O., Alarcon, G. S., Pratt, P. W. & Reveille, J. D. Most African-American patients with rheumatoid arthritis do not have the rheumatoid antigenic determinant (epitope). Ann. Intern. Med. 123, 181–187 (1995).
Hughes, L. B., Morrison, D., Kelley, J. M., Padilla, M. A., Vaughan, L. K. & Westfall, A. O. et al. The HLA-DRB1 shared epitope is associated with susceptibility to rheumatoid arthritis in African Americans through European genetic admixture. Arthritis Rheum. 58, 349–358 (2008).
Lundstrom, E., Kallberg, H., Smolnikova, M., Ding, B., Ronnelid, J. & Alfredsson, L. et al. Opposing effects of HLA-DRB1*13 alleles on the risk of developing anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis. Arthritis Rheum. 60, 924–930 (2009).
Okada, Y., Kim, K., Han, B., Pillai, N. E., Ong, R. T. & Saw, W. Y. et al. Risk for ACPA-positive rheumatoid arthritis is driven by shared HLA amino acid polymorphisms in Asian and European populations. Hum. Mol. Genet. 23, 6916–6926 (2014).
Reynolds, R. J., Ahmed, A. F., Danila, M. I., Hughes, L. B., Gregersen, P. K. & Raychaudhuri, S. et al. HLA-DRB1-associated rheumatoid arthritis risk at multiple levels in African Americans: hierarchical classification systems, amino acid positions, and residues. Arthritis Rheumatol. 66, 3274–3282 (2014).
Hung, S. I., Chung, W. H., Liou, L. B., Chu, C. C., Lin, M. & Huang, H. P. et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc. Natl Acad. Sci. USA 102, 4134–4139 (2005).
Mallal, S., Nolan, D., Witt, C., Masel, G., Martin, A. M. & Moore, C. et al. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet 359, 727–732 (2002).
Hetherington, S., Hughes, A. R., Mosteller, M., Shortino, D., Baker, K. L. & Spreen, W. et al. Genetic variations in HLA-B region and hypersensitivity reactions to abacavir. Lancet 359, 1121–1122 (2002).
Chung, W. H., Hung, S. I., Hong, H. S., Hsih, M. S., Yang, L. C. & Ho, H. C. et al. Medical genetics: a marker for Stevens-Johnson syndrome. Nature 428, 486 (2004).
Ozeki, T., Mushiroda, T., Yowang, A., Takahashi, A., Kubo, M. & Shirakata, Y. et al. Genome-wide association study identifies HLA-A*3101 allele as a genetic risk factor for carbamazepine-induced cutaneous adverse drug reactions in Japanese population. Hum. Mol. Genet. 20, 1034–1041 (2011).
McCormack, M., Alfirevic, A., Bourgeois, S., Farrell, J. J., Kasperaviciute, D. & Carrington, M. et al. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. N. Engl. J. Med. 364, 1134–1143 (2011).
Shirato, S., Kagaya, F., Suzuki, Y. & Joukou, S. Stevens-Johnson syndrome induced by methazolamide treatment. Arch. Ophthalmol. 115, 550–553 (1997).
Tamai, H., Sudo, T., Kimura, A., Mukuta, T., Matsubayashi, S. & Kuma, K. et al. Association between the DRB1*08032 histocompatibility antigen and methimazole-induced agranulocytosis in Japanese patients with Graves disease. Ann. Intern. Med. 124, 490–494 (1996).
Hirata, K., Takagi, H., Yamamoto, M., Matsumoto, T., Nishiya, T. & Mori, K. et al. Ticlopidine-induced hepatotoxicity is associated with specific human leukocyte antigen genomic subtypes in Japanese patients: a preliminary case-control study. Pharmacogenomics J. 8, 29–33 (2008).
Kurosaki, M., Takagi, H. & Mori, M. HLA-A33/B44/DR6 is highly related to intrahepatic cholestasis induced by tiopronin. Dig. Dis. Sci. 45, 1103–1108 (2000).
Daly, A. K., Donaldson, P. T., Bhatnagar, P., Shen, Y., Pe'er, I. & Floratos, A. et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat. Genet. 41, 816–819 (2009).
Hautekeete, M. L., Horsmans, Y., Van Waeyenberge, C., Demanet, C., Henrion, J. & Verbist, L. et al. HLA association of amoxicillin-clavulanate—induced hepatitis. Gastroenterology 117, 1181–1186 (1999).
Mammen, A. L., Gaudet, D., Brisson, D., Christopher-Stine, L., Lloyd, T. E. & Leffell, M. S. et al. Increased frequency of DRB1*11:01 in anti-hydroxymethylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Care Res. (Hoboken) 64, 1233–1237 (2012).
Wei, C. Y., Chung, W. H., Huang, H. W., Chen, Y. T. & Hung, S. I. Direct interaction between HLA-B and carbamazepine activates T cells in patients with Stevens-Johnson syndrome. J. Allergy Clin. Immunol. 129, 1562–1569 (2012).
Illing, P. T., Vivian, J. P., Dudek, N. L., Kostenko, L., Chen, Z. & Bharadwaj, M. et al. Immune self-reactivity triggered by drug-modified HLA-peptide repertoire. Nature 486, 554–558 (2012).
Maddocks, J. L. & Slater, D. N. Toxic epidermal necrolysis, agranulocytosis and erythroid hypoplasia associated with sulphasalazine. J. R. Soc. Med. 73, 587–588 (1980).
Balci, D. D., Peker, E., Duran, N. & Dogramaci, C. A. Sulfasalazine-induced hypersensitivity syndrome in a 15-year-old boy associated with human herpesvirus-6 reactivation. Cutan. Ocul. Toxicol. 28, 45–47 (2009).
Tremblay, L., Pineton de Chambrun, G., De Vroey, B., Lavogiez, C., Delaporte, E. & Colombel, J. F. et al. Stevens-Johnson syndrome with sulfasalazine treatment: report of two cases. J. Crohns Colitis 5, 457–460 (2011).
Jeung, Y. J., Lee, J. Y., Oh, M. J., Choi, D. C. & Lee, B. J. Comparison of the causes and clinical features of drug rash with eosinophilia and systemic symptoms and stevens-johnson syndrome. Allergy Asthma Immunol. Res. 2, 123–126 (2010).
Hall, C. L., Jawad, S., Harrison, P. R., MacKenzie, J. C., Bacon, P. A. & Klouda, P. T. et al. Natural course of penicillamine nephropathy: a long term study of 33 patients. Br. Med. J. 296, 1083–1086 (1988).
Hall, C. L., Fothergill, N. J., Blackwell, M. M., Harrison, P. R., MacKenzie, J. C. & MacIver, A. G. The natural course of gold nephropathy: long term study of 21 patients. Br Med J 295, 745–748 (1987).
Obayashi, M., Uzu, T., Harada, T., Yamato, M., Takahara, K. & Yamauchi, A. Clinical course of bucillamine-induced nephropathy in patients with rheumatoid arthritis. Clin. Exp. Nephrol. 7, 275–278 (2003).
Ueda, S., Wakashin, M., Wakashin, Y., Yoshida, H., Iesato, K. & Mori, T. et al. Experimental gold nephropathy in guinea pigs: detection of autoantibodies to renal tubular antigens. Kidney Int. 29, 539–548 (1986).
Smith, W. & Ball, G. V. Lung injury due to gold treatment. Arthritis Rheum. 23, 351–354 (1980).
Shidara, K., Hoshi, D., Inoue, E., Yamada, T., Nakajima, A. & Taniguchi, A. et al. Incidence of and risk factors for interstitial pneumonia in patients with rheumatoid arthritis in a large Japanese observational cohort, IORRA. Mod. Rheumatol. 20, 280–286 (2010).
Sawada, T., Inokuma, S., Sato, T., Otsuka, T., Saeki, Y. & Takeuchi, T. et al. Leflunomide-induced interstitial lung disease: prevalence and risk factors in Japanese patients with rheumatoid arthritis. Rheumatology (Oxford) 48, 1069–1072 (2009).
Koike, R., Tanaka, M., Komano, Y., Sakai, F., Sugiyama, H. & Nanki, T. et al. Tacrolimus-induced pulmonary injury in rheumatoid arthritis patients. Pulm. Pharmacol. Ther. 24, 401–406 (2011).
Furukawa, H., Oka, S., Shimada, K. Rheumatoid Arthritis-Interstitial Lung Disease Study Consortium, Tsuchiya, N. & Tohma, S. HLA-A*31:01 and methotrexate-induced interstitial lung aisease in Japanese rheumatoid arthritis patients: A multi-drug hypersensitivity marker? Ann. Rheum. Dis. 72, 153–155 (2013).
Speerstra, F., Reekers, P., van de Putte, L. B., Vandenbroucke, J. P., Rasker, J. J. & de Rooij, D. J. HLA-DR antigens and proteinuria induced by aurothioglucose and D-penicillamine in patients with rheumatoid arthritis. J. Rheumatol. 10, 948–953 (1983).
Furukawa, H., Oka, S., Shimada, K., Sugii, S., Hashimoto, A. & Komiya, A. et al. HLA-DRB1*08:02 is associated with bucillamine-induced proteinuria in Japanese rheumatoid arthritis patients. Biomark Insights 9, 23–28 (2014).
Tishler, M., Golbrut, B., Shoenfeld, Y. & Yaron, M. Anti-Ro(SSA) antibodies in patients with rheumatoid arthritis—a possible marker for gold induced side effects. J. Rheumatol. 21, 1040–1042 (1994).
Tishler, M., Paran, D. & Yaron, M. Allergic disorders in primary Sjogren's syndrome. Scand. J. Rheumatol. 27, 166–169 (1998).
Cruz-Tapias, P., Rojas-Villarraga, A., Maier-Moore, S. & Anaya, J. M. HLA and Sjogren's syndrome susceptibility. A meta-analysis of worldwide studies. Autoimmun. Rev. 11, 281–287 (2012).
Pachoula-Papasteriades, C., Boki, K., Varla-Leftherioti, M., Kappos-Rigatou, I., Fostiropoulos, G. & Economidou, J. HLA-A,-B, and -DR antigens in relation to gold and D-penicillamine toxicity in Greek patients with RA. Dis. Markers 4, 35–41 (1986).
Doyle, A. C. A Study in Scarlet. Beeton's Christmas Annual 28, 1–95 (1887).
Acknowledgements
We thank Ms Mayumi Yokoyama (Clinical Research Center for Allergy and Rheumatology, Sagamihara Hospital) and Ms Tomomi Hanawa (Clinical Research Center for Allergy and Rheumatology, Sagamihara Hospital) for secretarial assistance. The study at the Sagamihara Hospital was supported by Grants-in-Aid for Scientific Research (B, C; 26293123, 22591090) and for Young Scientists (B; 24791018) from the Japan Society for the Promotion of Science, Health and Labour Science Research Grants from the Ministry of Health, Labour, and Welfare of Japan, Grants-in-Aid for Clinical Research from the National Hospital Organization, Research Grants from Daiwa Securities Health Foundation, Research Grants from Japan Research Foundation for Clinical Pharmacology, Research Grants from The Nakatomi Foundation, Research Grants from Takeda Science Foundation, Research Grants from Mitsui Sumitomo Insurance Welfare Foundation, Research Grants from Kato Memorial Trust for Nambyo Research and research grants from the following pharmaceutical companies: Abbott Japan Co., Ltd., Astellas Pharma Inc., Chugai Pharmaceutical Co., Ltd., Eisai Co., Ltd., Mitsuibishi Tanabe Pharma Corporation, Merck Sharp and Dohme Inc., Pfizer Japan Inc., Takeda Pharmaceutical Company Limited, Teijin Pharma Limited. The funders had no role in study design, data collection and analysis, decision to publish, or preparing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
HF has the following conflicts, and the following funders are supported wholly or in part by the indicated pharmaceutical companies. The Japan Research Foundation for Clinical Pharmacology is run by Daiichi Sankyo, the Takeda Science Foundation is supported by an endowment from Takeda Pharmaceutical Company and the Nakatomi Foundation was established by Hisamitsu Pharmaceutical Co., Inc. The Daiwa Securities Health Foundation was established by the Daiwa Securities Group Inc. and Mitsui Sumitomo Insurance Welfare Foundation was established by the Mitsui Sumitomo Insurance Co., Ltd. HF received honoraria from Ajinomoto Co., Inc., Daiichi Sankyo Co., Ltd., Dainippon Sumitomo Pharma Co., Ltd., Pfizer Japan Inc. and Takeda Pharmaceutical Company. ST was supported by research grants from nine pharmaceutical companies: Abbott Japan Co., Ltd., Astellas Pharma Inc., Chugai Pharmaceutical Co., Ltd., Eisai Co., Ltd., Mitsubishi Tanabe Pharma Corporation, Merck Sharp and Dohme Inc., and Pfizer Japan Inc., Takeda Pharmaceutical Company Limited, Teijin Pharma Limited. ST received honoraria from Asahi Kasei Pharma Corporation, Astellas Pharma Inc., AbbVie GK., Chugai Pharmaceutical Co., Ltd., Ono Pharmaceutical Co., Ltd., Mitsubishi Tanabe Pharma Corporation and Pfizer Japan Inc. The remaining authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Furukawa, H., Oka, S., Shimada, K. et al. Human leukocyte antigen polymorphisms and personalized medicine for rheumatoid arthritis. J Hum Genet 60, 691–696 (2015). https://doi.org/10.1038/jhg.2015.36
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.36
This article is cited by
-
Management of rheumatoid arthritis in clinical practice using treat-to-target strategy: Where do we stand in the multi-ethnic Malaysia population?
Rheumatology International (2017)
-
Associations of human leukocyte antigens with autoimmune diseases: challenges in identifying the mechanism
Journal of Human Genetics (2015)
-
Human immune system diversity and its implications in diseases
Journal of Human Genetics (2015)
