
Abstract
Multiple pterygium syndrome (MPS) is an autosomal recessively inherited condition that becomes evident before birth, with pterygium at multiple joints and akinesia. There are two forms of this syndrome that are differentiated by clinical severity: the milder form, Escobar type (OMIM#265000), and the more severe form, lethal type (OMIM#253290). Mutations in CHRNG, which encode the acetylcholine receptor gamma subunit, cause most cases of MPS. Here, we present three patients from two unrelated families showing multiple joint contractures in both the upper and lower limbs. High-arched palates with malocclusion, short neck and micrognathia were observed in all patients. Peripheral blood karyotypes were normal. Whole-exome sequencing analysis of the patients’ genomes led to the discovery of identical missense (p.Pro143Arg) and frameshift deletion variants (p.Pro251fs*45) on CHRNG. These were rare cases of congenital arthrogryposis multiplex related to novel recessive CHRNG variants in two Korean kindred without apparent relatedness.
Similar content being viewed by others

Introduction
An acetylcholine receptor (AChR) is an integral membrane protein that is involved in a wide range of physiological signaling pathways by responding to acetylcholine.1 The nicotinic AChR includes muscle-type and neuronal-type. The binding of acetylcholine to muscle-type nicotinic AChRs at nerve–muscle synapses causes a conformational change in the receptor’s nonselective cation pore, which results in a rapid increase in the cellular permeability of cations and a generation of excitation of the muscle cell leading to a muscular contraction. Mutations of the gamma subunit gene of AChR, CHRNG (cholinergic receptor, nicotinic, gamma) represent a major cause of multiple pterygium syndrome (MPS), a form of arthrogryposis multiplex congenita that is a rare autosomal recessive condition characterized by pterygia, congenital contractures of the multiple joints and scoliosis. Other variable features include intrauterine death, congenital respiratory distress, short stature, ptosis, low-set ears and cryptorchidism in males.2 However, the impact of the CHRNG mutational spectrum on its clinical manifestation remains unclear.3, 4
Using whole-exome sequencing (WES) technique, we demonstrate that two independent Korean kindred of congenital arthrogryposis multiplex without typical features of Escobar syndrome showing severe pterygium are caused by rare, but identical, mutations in CHRNG.
Materials and methods
Subjects
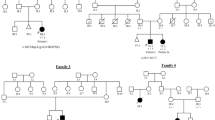
The first patient is a 5-year-old male (MPS1-1; Figure 1a) who visited an outpatient clinic for the evaluation of scoliosis and gait disturbance. He had pre- and perinatal problems including decreased fetal movement and multiple joint contractures noted at birth. However, he did not show severe intrauterine growth retardation and hydrops fetalis. The examination revealed both knees flexion contracture, vertical tali, triangular-shaped mouth, low-set ear, camptodactyly of multiple fingers and thoracolumbar scoliois. His height and weight were −4.4 s.d. and −2.5 s.d., respectively. Despite knee flexion contractures and vertical tali, he was able to walk independently and was of normal intelligence. From his family history (Figure 1e), his elder brother also had contractures involving multiple joints and died at 6 months of age because of respiratory infection. An electrodiagnostic study did not reveal peripheral neuromuscular causes.

Clinical representation of the affected individuals and compound heterozygous variants of CHRNG in this family. (a) Photograph of MPS1-1 shows both knee flexion contracture and rocker bottom foot deformity. (b) Camptodactyly with ulnar deviation of the second to fifth fingers on both hands are noted in MPS2-1. (c and d) A small mouth, micrognathia, a short neck and sloping shoulders are shown in facial photographs of MPS2-1 (c) and MPS2-2 (d). (e) Pedigrees of the families carrying recessive CHRNG mutations. Affected and unaffected individuals are denoted by black and white symbols, respectively. CHRNG alleles are represented by ‘+’ (wild-type), ‘P143R’ (p.Pro143Arg, missense mutation) and ‘P251fs’ (p.Pro251fs*45, frameshift deletion). (f) Amino-acid conservation of the CHRNG Pro143 residue in orthologs from different vertebrate species. B.t., Bostaurus; D.r., Danio rerio; G.g., Gallus gallus; H.s., Homo sapiens; M.m., Mus musculus and X.t., Xenopus tropicalis. A full color version of this figure is available at the Journal of Human Genetics journal online.
The second patient is a 12-year-old female (MPS2-1; Figure 1c) who was referred for the evaluation of congenital multiple joint contractures. Fetal movement was decreased during her pregnancy, and multiple joint contractures noted just after birth. Radiological examination revealed vertical tali in both feet, acetabular dysplasia of both hip joints, contractures of both knees and camptodactyly with ulnar deviation of fingers on both hands (Figure 1b). Contracture-release operations of the fingers and knees, and open reduction and realignment of tarsal bones were performed. Her height and weight were −0.7 s.d. and +3.9 s.d., respectively. Although she could walk alone without support and her intelligence was normal, she showed a small mouth, high-arched palate with malocclusion, micrognathia, a short neck and sloping shoulders.
The younger sister of patient 2 (MPS2-2; Figure 1d) had similar symptoms and signs of multiple joints. Prenatal findings were unremarkable except decreased fetal movement. She underwent surgeries for congenital vertical tali, camptodactyly of multiple fingers and contractures of both knee joints. She also showed bilateral ptosis, a small mouth, high-arched palate with malocclusion, micrognathia and a short neck with sloping shoulders. Her height and weight were −0.8 s.d. and +3.9 s.d., respectively. She could walk independently and showed normal intellectual development.
Whole-exome sequencing
Written informed consent was obtained from the patients and their parents to participate in this study. An Institutional Review Board approved this study. To understand the genetic basis of the clinical manifestations described above, we performed WES of two patients from each family (MPS1-1 and MPS2-1; Figure 1e). The experimental processes were as described previously, with minor modifications (Supplementary Table S1).5, 6 On the basis of the pedigree analysis, a recessive inheritance pattern was assumed. All the called variants were filtered according to the predicted effect on the protein and population frequencies (Table 1). Then, the causative variants were expected to be damaging based on conservation scores, such as PhyloP, and functional prediction software, such as PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://sift.jcvi.org/).7, 8, 9
Results
It is notable that the two families carried the same CHRNG variants—one missense (p.Pro143Arg) and one frameshift deletion (p.Pro251fs*45) (Figure 1e). The variants were confirmed by Sanger sequencing (Supplementary Figure S1), and are not listed in the 1000 genomes or the National Heart, Lung and Blood Institute whole-exome cohort. In the Korean population, the p.Pro143Arg mutation was found at an allele frequency of 1/1142 (~0.1%), whereas the frameshift mutation was not discovered in 1142 chromosomes. The two variants found in our patients were independently inherited from their parents. Our two families were not related, as demonstrated by rare genomic variant sharing analysis (Supplementary Figure S2). However, they shared a short haplotype (~17 kb) surrounding the CHRNG locus, commonly discovered in the Korean population (38/229=16.6%; data not shown), which implies either a relatively recent emergence of the mutations, followed by familial divergence, or independent generation of the mutations in two different lineages. The p.Pro143Arg variant is located in an extracellular domain of the protein and was a well-conserved amino-acid residue during evolution, which made it possible to assume functional implications based on change (Figure 1f and Supplementary Figure S3). The frameshift variant, a dinucleotide deletion, is expected to cause mis-translation of following 44 amino acids and finally a premature truncation at the 295th position, right after the second transmembrane domain (Supplementary Figure S3).
Discussion
Mutations of the AChR pathway genes (CHRNA1, CHRNB1, CHRND, CHRNG and RAPSN) are known to be related with various forms of fetal akinesia deformation sequence showing fetal movement restriction, intrauterine growth retardation, fetal hydrops, micrognathia and multiple joint contractures with or without pterygia.10
Among these genes, CHRNG encodes the gamma subunit of the embryonic AChR, and the gamma subunit has roles not only in neuromuscular signal transduction, but also in neuromuscular junction formation during the fetal period.11 CHRNG mutations are known to be the cause of the non-lethal Escobar variant or the lethal form of MPS, and have been reported to be correlated with the fetal akinesia deformation sequence without pterygia (Supplementary Table S2). Vogt et al.4 reported that almost all MPS patients with CHRNG mutations showed pterygia, which rendered it possible to associate the CHRNG mutations with pterygia. Although fetal conditions of our patients were not severe and pterygia were not evident, congenital arthrogryposis multiplex shown in our patients can be understood as a manifestation of fetal akinesia deformation sequence in a broader context.
In this case, the gamma subunit of the AChR, which has a role in the muscle-relaxant effect, was mutated. Although the two unrelated patients were not related, it is likely that the mutant alleles arose in founders and propagated in the Korean population as rare forms. The discovery of identical CHRNG variants raises awareness among anesthesiologists if the patient was to be subjected to an operation for joint deformity. Moreover, the pleiotropic effect of CHRNG should be noted; it is also associated with congenital diaphragmatic muscle weakness, pulmonary hypoplasia and intraspinal abnormalities, such as syringomyelia or tethered cord syndrome. Therefore, it is worth considering these genetic results when planning a surgical operation or a rehabilitation program.
We report identical compound heterozygous variants using WES, one missense mutation (p.Pro143Arg) and one frameshift deletion (p.Pro251fs*45), on CHRNG discovered in two unrelated Korean patients with a similar clinical presentation. These mutations are reported for the first time as cause of congenital arthrogryposis multiplex associated with the AChR pathway. Considering that our two families were unrelated, these two novel variants might represent a founder mutation in the Korean population, and understanding these mutations may provide better treatment and follow-up plans.
References
Karlin, A . Emerging structure of the nicotinic acetylcholine receptors. Nat. Rev. Neurosci. 3, 102–114 (2002).
Morgan, N. V ., Brueton, L. A ., Cox, P ., Greally, M. T ., Tolmie, J . & Pasha, S . et al. Mutations in the embryonal subunit of the acetylcholine receptor (CHRNG) cause lethal and Escobar variants of multiple pterygium syndrome. Am. J. Hum. Genet. 79, 390–395 (2006).
Hoffmann, K ., Muller, J. S ., Stricker, S ., Megarbane, A ., Rajab, A . & Lindner, T. H . et al. Escobar syndrome is a prenatal myasthenia caused by disruption of the acetylcholine receptor fetal gamma subunit. Am. J. Hum. Genet. 79, 303–312 (2006).
Vogt, J ., Morgan, N. V ., Rehal, P ., Faivre, L ., Brueton, L. A . & Becker, K . et al. CHRNG genotype-phenotype correlations in the multiple pterygium syndromes. J. Med. Genet. 49, 21–26 (2012).
Choi, M ., Scholl, U. I ., Ji, W ., Liu, T ., Tikhonova, I. R . & Zumbo, P . et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl Acad. Sci. USA 106, 19096–19101 (2009).
Goh, G . & Choi, M . Application of whole exome sequencing to identify disease-causing variants in inherited human diseases. Genomics Inform. 10, 214–219 (2012).
Pollard, K. S ., Hubisz, M. J . & Siepel, A . Detection of non-neutral substitution rates on mammalian phylogenies. Genome Res. 20, 110–121 (2010).
Ng, P. C . & Henikoff, S . SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814 (2003).
Adzhubei, I. A ., Schmidt, S ., Peshkin, L ., Ramensky, V. E ., Gerasimova, A . & Bork, P . et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Michalk, A ., Stricker, S ., Becker, J ., Rupps, R ., Pantzar, T . & Miertus, J . et al. Acetylcholine receptor pathway mutations explain various fetal akinesia deformation sequence disorders. Am. J. Hum. Genet. 82, 464–476 (2008).
Koenen, M ., Peter, C ., Villarroel, A ., Witzemann, V . & Sakmann, B . Acetylcholine receptor channel subtype directs the innervation pattern of skeletal muscle. EMBO Rep. 6, 570–576 (2005).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Seo, J., Choi, IH., Lee, J. et al. Rare cases of congenital arthrogryposis multiplex caused by novel recurrent CHRNG mutations. J Hum Genet 60, 213–215 (2015). https://doi.org/10.1038/jhg.2015.2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.2

{kind=link}
{kind=link}
{kind=link}