
Abstract
Recently, de novo mutations in TBL1XR1 were found in two patients with autism spectrum disorders. Here, we report on a Japanese girl presenting with West syndrome, Rett syndrome-like and autistic features. Her initial development was normal until she developed a series of spasms at 5 months of age. Electroencephalogram at 7 months showed a pattern of hypsarrhythmia, which led to a diagnosis of West syndrome. Stereotypic hand movements appeared at 8 months of age, and autistic features such as deficits in communication, hyperactivity and excitability were observed later, at 4 years and 9 months. Whole exome sequencing of the patient and her parents revealed a de novo TBL1XR1 mutation [c.209 G>A (p.Gly70Asp)] occurring at an evolutionarily conserved amino acid in an F-box-like domain. Our report expands the clinical spectrum of TBL1XR1 mutations to West syndrome with Rett-like features, together with autistic features.
Similar content being viewed by others


Main
TBL1XR1 at 3q26.32 encodes transducin β-like 1 X-linked receptor 1, a co-repressor of nuclear hormone transcription factors that is required for β-catenin–Tcf-mediated Wnt signaling.1, 2, 3 Recently, two de novo TBL1XR1 mutations were found in 2 of 2446 patients with autism spectrum disorders, suggesting an association between TBL1XR1 mutations and autism.4, 5 Here, we present a third case with a de novo TBL1XR1 mutation, showing West syndrome, Rett-like and autistic features.
Case report
This report concerns a 5-year-old girl who is the offspring of unrelated healthy Japanese parents. She was born at 39 weeks of gestation without asphyxia after an uneventful pregnancy. Her birth weight, birth length and head circumference were 3088 g (+0.3 standard deviation (s.d.)), 51.1 cm (+1.0 s.d.) and 34.0 cm (+0.5 s.d.), respectively. She showed social smiling and head control at 3 and 4 months of age, respectively. Then at 5 months she developed a series of spasms occurring 5–6 times a day, when her head control became unstable. Electroencephalography at 7 months of age showed hypsarrhythmia patterns (Figure 1a), which led to a diagnosis of West syndrome. Brain magnetic resonance imaging showed no structural brain anomalies (Figures 1a and c). Administration of adrenocorticotropic hormone therapy only temporarily reduced the frequency of spasms.

Electroencephalogram (EEG) and brain magnetic resonance imaging in the patient at 7 months of age. (a) Interictal EEG showed high-amplitude multifocal spikes with irregular slow waves consistent with a finding of hypsarrhythmia. T2-weighted axial images through the basal ganglia (b) and the cerebellum (c) showed no abnormalities.
On examination at 7 months of age, the patient’s weight, height and head circumference were 8720 g (+1.2 s.d.), 69.5 cm (+0.9 s.d.) and 42 cm (−0.6 s.d.), respectively. Mild dysmorphic features were observed, including a long palpebral fissure, thick eyebrows and downturned corners of the mouth. The patient showed no eye fixation and pursuit, as well as no social smile. Her muscle tone was mildly hypotonic. Despite being given extensive treatments including adrenocorticotropic hormone therapy in combination with valproic acid, nitrazepam, vitamin B6, topiramate, clonazepam and clobazam, she continued to exhibit frequent subtle tonic seizures with eyelid opening. At 8 months of age, stereotypical hand movements appeared that resembled hand-washing.
Laboratory examination revealed elevated serum levels of several components: lactic acid (39.9 mg dl−1, compared with a normal range of 5.0–20.0 mg dl−1), pyruvate (2.79 mg dl−1, normal range 0.3–0.9 mg dl−1) and alanine (1447 nmol ml−1, normal range 180–470 nmol ml−1). However, following vitamin B1 treatment, both pyruvate and alanine serum levels returned to normal. The levels of lactic acid and pyruvate in cerebrospinal fluid appeared normal (14.6 and 0.65 mg dl−1, respectively). Respiratory-chain enzymes, using muscle homogenates and fibroblasts, and mitochondrial DNA sequence analysis were all normal, and pathological examination of muscle specimens revealed no specific findings. Repeated examination of both serum lactic acid and pyruvate levels also showed no abnormalities.
After contracting a fever at the age of 1 year and 10 months, her seizures were controlled using a combination therapy of topiramate, valproic acid, clobazam and vitamin B1. However, the patient still could not speak any meaningful words at 4 years and 9 months of age but could walk with support; she had a developmental quotient of 13. At this age she still showed stereotypic hand movements as well as autistic features such as deficits in communication, hyperactivity and excitability.
Results and discussion
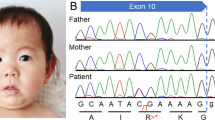
G-banded karyotyping was normal (46,XX). No pathological copy number aberrations were detected by the 2.7M Array (Affymetrix, Santa Clara, CA, USA). Whole exome sequencing of the patient and her parents was performed. Genomic DNA of blood leukocytes was captured using the SeqCap EZ Exome Library v2.0 (Roche NimbleGen, Madison, WI, USA), and sequenced with on HiSeq2000 (Illumina, San Diego, CA, USA). Variants were detected as previously described.6 Variants with a Phred-like consensus quality score of >100 were considered as candidate variants. We found a total of four de novo candidate variants, in which two mutations were further validated as de novo by Sanger sequencing: SELPLG NM_001206609.1:c.794C>T (p.Thr265Met) and TBL1XR1 NM_024665.4:c.209G>A (p.Gly70Asp). The other two variants were transmitted from her mother, demonstrating that the two variants were falsely uncalled in the mother. Neither of the two de novo mutations was found in the 6500 National Heart, Lung, and Blood Institute exomes nor in our 575 in-house control exomes. Both mutations were predicted as damaging by SIFT and PolyPhen2. However, MutationTaster classified only p.Gly70Asp in TBL1XR1 as damaging whereas p.Thr265Met in SELPLG was predicted as a polymorphism. We found no recessive mutations in known early-onset epileptic encephalopathy genes including SLC25A22, PNPO, PNKP and PLCB1.7 All experimental protocols used were approved by the Institutional Review Board of Yokohama City University School of Medicine.
SELPLG encodes P-selectin glycoprotein ligand 1, for which a knock-out mouse study showed that Selplg is required for leukocyte adhesion and rolling.8, 9 No neurological abnormalities were reported, suggesting that the SELPLG mutation is less likely to be involved in the phenotype of this patient.
TBL1XR1, also denoted as TBLR1, is required for β-catenin–Tcf-mediated Wnt signaling.1, 2 Mutations in TCF4, an essential mediator of Wnt signaling, have been shown to cause Pitt–Hopkins Syndrome, which presents with severe intellectual disability, seizures and stereotypic movements.10, 11 This suggests that the β-catenin–Tcf-mediated Wnt pathway of signaling is essential for normal brain function. Moreover, two de novo TBL1XR1 mutations (p.Leu282Pro and p.Ile397SerfsX19) were found in 2 of 2446 patients with autism spectrum disorders.4, 5 In our case, the p.Gly70Asp mutation occurred in an evolutionarily conserved amino acid within an F-box-like domain (Figure 2). Indeed, the F-box-like domain of TBLR1 (TBL1XR1) is essential for a high affinity interaction between TBL1XR1 and SMRT, a co-repressor of nuclear hormone receptors.3 This implies that p.Gly70Asp may affect this interaction. Therefore, the evidence suggests that the p.Gly70Asp mutation may cause a West syndrome phenotype with Rett-like and autistic features.

De novo TBL1XR1 mutation. The c.209G>A (p.Gly70Asp) mutation occurred de novo at an evolutionarily conserved amino acid in an F-box-like domain. Multiple amino-acid sequences of TBL1XR1 proteins were aligned with tools available on the CLUSTALW web site. Two previously reported mutations (p.Leu282Pro and p.Ile397SefsX19) are highlighted in red. A full color version of this figure is available at the Journal of Human Genetics journal online.
The role of TBL1XR1 mutations was also investigated in 280 epileptic patients. High-resolution melting analysis revealed that three rare missense variants (p.Ala116Ser, p.Gly405Glu and p.Asn407Ser) were present in three patients. Since they were predicted as benign by PolyPhen-2, these mutations are unlikely to be pathogenic. These data suggest that TBL1XR1 mutations are rarely involved in epileptic patients.
In conclusion, we describe a Japanese girl with a de novo TBL1XR1 mutation that is predicted as pathogenic. Our report suggests that the clinical spectrum of TBL1XR1 mutations includes autistic features as a core phenotype, as well as presenting with West syndrome and Rett-like features.
References
Li, J. & Wang, C. Y. TBL1-TBLR1 and beta-catenin recruit each other to Wnt target-gene promoter for transcription activation and oncogenesis. Nat. Cell Biol. 10, 160–169 (2008).
Choi, H. K., Choi, K. C., Yoo, J. Y., Song, M., Ko, S. J., Kim, C. H. et al. Reversible SUMOylation of TBL1-TBLR1 regulates beta-catenin-mediated Wnt signaling. Mol. Cell 43, 203–216 (2011).
Zhang, X. M., Chang, Q., Zeng, L., Gu, J., Brown, S. & Basch, R. S. TBLR1 regulates the expression of nuclear hormone receptor co-repressors. BMC Cell Biol. 7, 31 (2006).
O'Roak, B. J., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B. P. et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250 (2012).
O'Roak, B. J., Vives, L., Fu, W., Egertson, J. D., Stanaway, I. B., Phelps, I. G. et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–1622 (2012).
Walsh, T., Lee, M. K., Casadei, S., Thornton, A. M., Stray, S. M., Pennil, C. et al. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc. Natl Acad. Sci. USA 107, 12629–12633 (2010).
Mastrangelo, M. & Leuzzi, V. Genes of early-onset epileptic encephalopathies: from genotype to phenotype. Pediatr. Neurol. 46, 24–31 (2012).
Yang, J., Hirata, T., Croce, K., Merrill-Skoloff, G., Tchernychev, B., Williams, E. et al. Targeted gene disruption demonstrates that P-selectin glycoprotein ligand 1 (PSGL-1) is required for P-selectin-mediated but not E-selectin-mediated neutrophil rolling and migration. J. Exp. Med. 190, 1769–1782 (1999).
Xia, L., Sperandio, M., Yago, T., McDaniel, J. M., Cummings, R. D., Pearson-White, S. et al. P-selectin glycoprotein ligand-1-deficient mice have impaired leukocyte tethering to E-selectin under flow. J. Clin. Invest. 109, 939–950 (2002).
Zweier, C., Peippo, M. M., Hoyer, J., Sousa, S., Bottani, A., Clayton-Smith, J. et al. Haploinsufficiency of TCF4 causes syndromal mental retardation with intermittent hyperventilation (Pitt-Hopkins syndrome). Am. J. Hum. Genet. 80, 994–1001 (2007).
Amiel, J., Rio, M., de Pontual, L., Redon, R., Malan, V., Boddaert, N. et al. Mutations in TCF4, encoding a class I basic helix-loop-helix transcription factor, are responsible for Pitt-Hopkins syndrome, a severe epileptic encephalopathy associated with autonomic dysfunction. Am. J. Hum. Genet. 80, 988–993 (2007).
Acknowledgements
We would like to thank the patient and her family for their participation in this study. We thank Kiyomi Nishiyama for her technical assistance. This study was supported by: the Japanese Ministry of Health, Labour, and Welfare; the Japan Society for the Promotion of Science (a Grant-in-Aid for Scientific Research (B) (25293085, 25293235) and a Grant-in-Aid for Scientific Research (A) (13313587)); the Takeda Science Foundation; the fund for Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems; the Strategic Research Program for Brain Sciences (11105137); and a Grant-in-Aid for Scientific Research on Innovative Areas (Transcription Cycle) from the Japanese Ministry of Education, Culture, Sports, Science, and Technology (12024421).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Saitsu, H., Tohyama, J., Walsh, T. et al. A girl with West syndrome and autistic features harboring a de novo TBL1XR1 mutation. J Hum Genet 59, 581–583 (2014). https://doi.org/10.1038/jhg.2014.71
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2014.71
This article is cited by
-
The spectrum of neurological presentation in individuals affected by TBL1XR1 gene defects
Orphanet Journal of Rare Diseases (2024)
-
Understanding the Landscape of X-linked Variants Causing Intellectual Disability in Females Through Extreme X Chromosome Inactivation Skewing
Molecular Neurobiology (2020)
-
Monogenic disorders that mimic the phenotype of Rett syndrome
neurogenetics (2018)
-
De novo non-synonymous TBL1XR1 mutation alters Wnt signaling activity
Scientific Reports (2017)
-
Transducin (β)-like 1 X-linked receptor 1 promotes gastric cancer progression via the ERK1/2 pathway
Oncogene (2017)
