
Abstract
Gitelman’s syndrome (GS) is a rare recessive disorder caused by mutations in the renal salt-handling genes SLC12A3 and CLCNKB. Our aim was to develop a next-generation sequencing (NGS) procedure for these genes based on two-tubes multiplex amplification of DNA pools and semiconductor sequencing with the Ion Torrent Personal Genome Machine (PGM). We created one pool with DNA from 20 GS patients previously Sanger sequenced for the coding exons of SLC12A3. A total of 13 mutations present in 11 of these patients were used as control variants to validate the NGS procedure. The full coding sequence of SLC12A3, CLCNKB and CLCNKA was amplified in only two Ampliseq tubes and processed and sequenced with the PGM. Large SLC12A3 and CLCNKB deletions were ascertained through multiplex ligation-dependent probe amplification in some patients. With the exception of the SLC12A3 exon 9, all the amplicons were successfully read and 12 of the 13 control variants were detected. The analysis of CLCNKB showed four putative mutations in the GS pool that were further assigned to specific patients. Two patients were heterozygous compounds for a single-nucleotide mutation and a large deletion at SLC12A3 or CLCNKB. We reported a NGS procedure that would facilitate the rapid and cost-effective large-scale screening of the three renal salt-handling genes. In addition to characterize the mutational spectrum of GS patients, the described procedure would facilitate the rapid and cost-effective screening of these genes at a population scale.
Similar content being viewed by others
Introduction
Gitelman’s syndrome (GS; OMIM 263800) is an autosomal recessive kidney tubular disease. Most of the GS cases have loss-of-function mutations in the SLC12A3 gene, which encodes the thiazide-sensitive Na–Cl cotransporter.1, 2, 3, 4 Mutations in the CLCNKB gene, which encodes the renal chloride channel ClC-Kb might also resemble manifestations of GS in some patients.5, 6 The Na–Cl cotransporter mediates the sodium and chloride reabsorption in the luminal membrane of the distal convoluted tubule, and its loss of function causes hypokalamic metabolic alkalosis secondary to sodium depletion and subsequent stimulation of the renin–angiotensin–aldosterone axis.7 Patients with GS exhibit a broad spectrum of symptoms, from asymptomatic subjects to severe cases of rhabdomyolysis, tetany and paralysis.8 No phenotype–genotype correlation has been demonstrated.
The SLC12A3 gene encompasses >100 kb and contains 26 coding exons. With exceptions (as the intron 9+1 G>T in gypsies), the same mutation has been found in only one or few patients.4, 9 In practical terms, this means that the 26 SLC12A3 exons (plus 2–5 intron-flanking nucleotides) should be sequenced in most GS patients. In addition, sequencing of the CLCNKB gene needs to be carried out in patients negative for SLC12A3 mutations. As a result of the large size of these genes, the Sanger-based sequencing of single exon-amplicons is labor-intensive and expensive and fully justifies the use of next-generation sequencing (NGS) technologies for the mutation screening of the GS-genes.
The Ion Torrent Personal Genome Machine (PGM) is a semiconductor (instead of optical) NGS technology.10, 11 The reported NGS procedures are based on the amplification of DNA fragments from single patients, followed by labeling of the whole amplicons with specific barcode primers and sequencing. Because each fragment can be recognized by the corresponding barcode, several patients can be sequenced in a single array. The amplification and sequencing of DNA pools from several individuals could reduce the cost and effort of handling large sample sets, but the nucleotide variants present in only one individual are diluted by the wild-type allele and this could result in a signal too low to be detected (false negatives).12
The purpose of this study was to develop a rapid and cost-effective procedure for sequencing SLC12A3, CLCNKB and CLCNKA (closely linked to CLCNKB) in pooled DNA samples.
Materials and methods
Patients and samples preparation
This study was approved by the Ethical Committee of Hospital Universitario Central de Asturias. A total of 20 patients diagnosed with GS and previously Sanger sequenced for the 26 SLC12A3-coding exons were recruited through the Paediatric Nephrology Department of Hospital Universitario Central de Asturias (Table 1). In 11 patients, at least one SLC12A3 mutation had been identified. In three cases, we confirmed the homozygous state (instead of a single-nucleotide mutation plus a large deletion of one or more exons) by sequencing the two parents (who were heterozygous carriers).
The DNA from each individual was obtained from leukocytes, resuspended in water and adjusted to 10 ng μl−1 using Real Time Taqman quantification with RNase P Detection Reagents (Life technologies, Carlsbad, CA, USA). One pool containing equimolecular quantities of each DNA was created. In this way, assuming equimolecular amounts of the 20 DNAs the frequency of reads for each unique allele (present in only one individual per pool) should be 2.5% (1/40 alleles).
Multiplex Ampliseq amplification
A multiplex amplification for the whole-coding sequence plus at least five intronic-flanking nucleotides of SLC12A3, CLCNKA and CLCNKB were designated online (Ion AmpliSeq Designer; https://www.ampliseq.com). A total of 68 primer-pairs in two tubes (34 different amplicons per tube) were provided by the manufacturer (Life technologies). The amplicons covered 100% of the CLCNKA/B and 99.91% of the SLC12A3-target sequences (Supplementary Table 1).
Libraries preparation and PGM sequencing
The Ampliseq reactions were processed with the Ion Ampliseq Library Kit (Life technologies) followed by Ion Torrent semiconductor sequencing. Briefly, 10 ng of the DNA pools were amplified with Ampliseq in two tubes, followed by the digestion of the primer sequences with FuPa reagent (Life technologies), ligation of a specific oligonucleotide (barcode) to each pool, purification by Agencourt AMPure XP reagent (Beckam Coulter, Brea, CA, USA), PCR with the adapters using Platinum PCR SuperMix High Fidelity enzyme (Invitrogen, Life Technologies), purification by Agencourt AMPure XP reagent, quantification (Agilent Bioanalyser Instrument, Agilent, Santa Clara, CA, USA and Qubit 2.0 Fluorometer, Life technologies) and dilution of the sample to a final concentration of 20 pM.
Libraries were amplified using the Ion PGM template OT2 200 Kit (Life technolgies) and the Ion One-Touch instrument (Life Technologies). Template-positive spheres were recovered using Dynabeads MyOne Streptavidin C1 beads (Life technolgies) and qualified using the Ion Sphere quality control assay and the Qubit 2.0 fluorometer (Life Technologies). Sphere particles were loaded in a 318 (1 Gb) semiconductor chip, and sequenced using the PGM 200 sequencing kit protocol in the Ion Torrent PGM. We used a 260-flow runs, which support a template read-length of ∼200 bp.
Data analysis
Data were processed using the Ion Torrent platform-specific pipeline software Torrent Suite v3.4.2 (Life technologies) to generate sequence reads filtered according to the pipeline software quality-controls and to remove poor signal reads. Reads assembling and variant identification were performed with the Variant Caller v3.4.51874 software using FastQ files containing sequence reads and the Ion Ampliseq Designer BED file software to map the amplicons. Integrative Genome Viewer was used for the analysis of depth coverage, sequences quality and variants identification. The variant caller algorithm was set at threshold frequency of 1% to identify the nucleotide variants.
Sanger sequencing
For each putative mutation in the pool, each DNA (used to create the pool) was amplified and sequenced with BigDye chemistry in an ABI3130xl equipment, to identify the individual who was the mutation carrier. Briefly, the exon containing the nucleotide variant was amplified with primers that matched the flanking introns and PCR fragments were purified and sequenced.
Multiplex ligation-dependent probe amplification analysis
In patients with only one SLC12A3-mutated allele, we determined the presence of SLC12A3 or CLCNKB copy number variants through multiplex ligation-dependent probe amplification assays (Salsa Kit P213, MRC Holland, Amsterdam, The Netherlands).
Results
NGS of the Gitelman’s pool
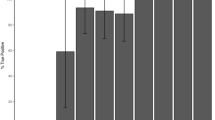
All the SLC12A3 Ampliseq amplicons rendered >2000 reads per base (>50 × coverage for each of the 20 DNAs in the pool), with the exception of one covering part of exon 9 (Figure 1). This amplicon failed in the four pools, and also in a run of four-sample pool performed in the low-capacity (1 Mb) 316 PGM array (data not shown). Thus, we concluded that the lack of sequence reads for this SLC12A3 amplicon was intrinsic to the Ampliseq design. In the GS patients pool, we identified several nucleotide variants in the three genes (Supplementary Table 2). At a 2.5% allele frequency threshold, six of the nine unique variants were not detected (Table 2). At a 1% threshold, eight were recognized as nucleotide variants, with approximately 50% of the nucleotide reads in each (forward/reverse) strand and no false-positive single-nucleotide variant calls (Supplementary Figure 1). The only non-detected control variant was the intron 9 c.1180 +1 G>T mutation, characteristic of the gypsy population. The visual inspection of the binary alignment map (BAN) file showed 0.8% of T-reads, but all on forward strands. The absence of nucleotide reads in reverse strands would thus explain the non-variant caller identification of the intron 9 +1 G>T mutation. Thus, a complete mutational screening of SLC12A3 would require the Sanger sequencing of exon 9.

Number of nucleotide reads for the SLC12A3 and CLCNKB amplicons in the Gitelman’s syndrome (GS) patients. The × 50 coverage (total reads=2000; mean reads per allele=50) is indicated.
Two of the 23 CLCNKB and four of the 23 CLCNKA amplicons gave depth coverage values below the standard 50 × (Figure 1). To reduce the risk for false negatives at these amplicons (truth nucleotide changes not recognized by the variant caller), we performed a visual inspection of the aligned reads. In the GS pools, we found several CLCNKB rare likely pathogenic variants that were determined in the 20 patients through Sanger sequencing of the corresponding exons (Table 3). One patient was homozygous for the missense change p.A204T, a mutation previously found in GS patients. One patient was compound heterozygous for this mutation and p.V170M (Supplementary Figure 2). In two patients (also SLC12A3-negatives), we only found one putative CLCNKB mutation (p.Y99H and p.E442G). All the CLCNKA variants in the GS pool were known polymorphisms.
In/dels idenfication
One of the main limitations of the PGM (and other NGS platforms) is the presence of false-variant calls, mainly in homopolymer regions ⩾four nucleotides and insertion/deletion of few nucleotides. The variant caller identified only two false-negative variants, both in homopolymer tracts of a CLCNKA intron (16354433 A>AT and 16354441 TC>C), and the only in/del control variant (p.K199 fs) was recognized.
SLC12A3 and CLCNKB gene rearrangements through multiplex ligation-dependent probe amplification
The multiplex ligation-dependent probe amplification was performed in the 13 patients in which only one (n=4) or no mutated (n=11) SLC12A3 allele was identified. One patient was heterozygous for a deletion of exons 4–5 of SLC12A3, and one patient should be heterozygous for a deletion of all the CLCNKB exons (Supplementary Figure 3). The parent positive for the single-nucleotide mutation was negative for the deletion. Thus, the two patients should be heterozygous compound for a deletion and a single-nucleotide mutation (Tables 1 and 3).
Discussion
NGS is a powerful tool to identify causative mutations in mendelian disorders, particularly useful when dealing with large genes and diseases with several causative genes. NGS has also facilitated the discovery of rare nucleotide variants at a population scale, that could be linked to the risk of common traits such as hypertension, blood lipid profile or type 2 diabetes. For most of the genes, the reported NGS procedures are based on the amplification of short (<200 bp) PCR fragments (amplicons) from single patients. Usually, each fragment is amplified in a single tube, all the amplicons from each patient are pooled and barcoded with an oligonucleotide before the creation of a library that is finally array-sequenced. The addition of a specific barcode makes possible to differentiate the sequences from many patients in a single-sequencing array.13, 14 In spite of these advantages for large-scale analysis (compared with the Sanger sequencing), the necessity of multiple PCRs and library preparations is costly in both, labor and money, when a laboratory requires the analysis of large amounts of individuals. A way to reduce these limitations should be the amplification of the target sequences in only two tubes.15, 16 In addition, the amplification of DNA pools from several individuals would avoid the necessity of multiple barcoding reducing the time and cost of large-scale analysis.12, 15, 17 Although rare sequence variants have been successfully identified through NGS of DNA pools, some authors suggested that amplification and barcoding of individual samples should be preferred to avoid false-positives.12 Thus, the ability to detect rare variants in a pool of DNAs is a critical issue that needs to be addressed.
The Ion Torrent PGM is a semiconductor (instead of optical)-based NGS platform that has been used to identify disease-causing mutations in some mendelian disorders.10, 11, 12, 13, 14, 15, 18 We designated a procedure to amplify the target amplicons of three genes that encoded salt-handling renal channels in only two tubes, and validated the coverage and accuracy of detecting rare variants in one pool of 20 patients. With this protocol, only one barcoded library per pool (instead of 20) should be necessary to identify the nucleotide variants in the 20 individuals. The amplification of many fragments per tube reduces the necessity of amplifying each fragment in a single tube, but at the cost of poor or no amplification for some of the fragments, mainly in GC-rich regions that are difficult to amplify even when the reaction tubes only contains the amplicon-specific primers (instead of a mixture of dozens of primer-pairs). Only one amplicon that encompasses most of the SLC12A3 exon 9 did not give nucleotide reads due to amplification failure. In this way, a full screening of the SLC12A3 gene would require the Sanger sequencing of exon 9 in each sample.
With the exception of the intron 9 +1 G>T variant, we identified all the SLC12A3 control variants present in the patients used to create the pool, with deep coverages >1%. One of the main limitations of the PGM (and other NGS platforms) is the presence of false-positives mainly in homopolymer regions ⩾four nucleotides and insertion/deletion of few nucleotides.11, 14 Although the only nucleotide deletion (p.K199 fs) was identified, we cannot exclude that other small in/dels are not identified when large amounts of patients are analyzed.
The NGS showed four putative CLCNKB mutations in the GS pool. After Sanger sequencing of the 20 patients, we found that the four were in patients negative for SLC12A3 mutations. One patient was homozygous for p.A204T, a mutation previously found in GS patients with early-onset symptoms.4 One patient was heterozygous compound for this mutation and p.V170M, an also reported mutation. The identification of the causative mutations in these cases illustrates the usefulness of our procedure to characterize the mutational spectrum of GS by the simultaneous sequencing of the SLC12A and CLCNKB. In patients heterozygous for p.Y99H and p.E442G (two non-reported mutations), a second single-nucleotide mutation was not found. We complemented the NGS with a search for large deletions in the SLC12A and CLCNKB genes, and identified two patients who should be heterozygous compound for SLC12A3 intron 21 splice defect andexon 4–5 deletion, and CLCNKB p.E442G and full exon deletion.
The NGS of DNA pools has been used by some authors for the mutation analysis of inherited disorders.17, 19 However, this approach has the risk of amplification failures for a particular DNA in a pool and in this case, the corresponding patient should be wrongly classified as non-mutation carrier. To reduce the risk for false negatives due to amplification failures, the pool was made with high-quality DNAs previously assayed and quantified through Taqman assays. The fact that all the control variants in all the SLC12A3 readable amplicons were detected suggested that our approach could be useful for the mutational screening of these genes. In addition, the power to detect rare mutations could be increased by sequencing overlapping population pools, where each individual occurs in two pools.20
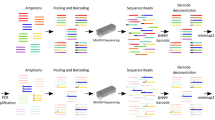
Finally, based on our experience we propose a flow chart for the NGS screening of GS patients (Figure 2). This could be specially useful to characterize large cohorts of patients without previous genetic study, or patients negative for SLC12A3 mutations but not sequenced for CLCNKB. All the putative mutations identified in a pool could be assigned to a specific patient through Sanger sequencing of the corresponding exon. The Sanger-based sequencing of all the coding exons from the two genes would require to amplify 46 fragments and 92 sequence reads (forward and reverse strands) per patient. In our case, a total of 520 PCR fragments and 1040 sequences should be necessary for the 20 patients. All the mutations identified in the pool were in 14 exons (10 in SLC12A3 and 4 in CLCNKB) and the number of amplifications and Sanger-sequencing reads could thus be reduced to 280 and 560, respectively. This represents an almost 50% reduction of the cost compared with the Sanger sequencing of all the patients.

Flow chart for the next-generation sequencing (NGS) of a DNA pool to characterize mutations in the SLC12A3 and CLCNKB, the main genes implicated in Gitelman’s syndrome.
In conclusion, we report the massive sequencing of three renal salt-handling genes through multiplex amplification of DNA pools and only two tubes per sample. The reported procedure would facilitate the rapid and cost-effective screening of these genes in large cohorts of GS patients.
References
Lemmink, H. H., Knoers, N. V., Karolyi, L., van Dijk, H., Niaudet, P., Antignac, C. et al. Novel mutations in the thiazidesensitive NaCl cotransporter gene in patients with Gitelman syndrome with predominant localization to the C-terminal domain. Kidney Int. 54, 720–730 (1998).
Simon, D. B., Nelson-Williams, C., Bia, M. J., Ellison, D., Karet, F. E., Molina, A. M. et al. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat. Genet. 12, 24–30 (1996).
Mastroianni, N., Bettinelli, A., Bianchetti, M., Colussi, G., De Fusco, M., Sereni, F. et al. Novel molecular variants of the Na-Cl cotransporter gene are responsible for Gitelman syndrome. Am. J. Hum. Genet. 59, 1019–1026 (1996).
Vargas-Poussou, R., Dahan, K., Kahila, D., Venisse, A., Riveira-Munoz, E., Debaix, H. et al. Spectrum of mutations in Gitelman syndrome. J. Am. Soc. Nephrol. 22, 693–703 (2011).
Zelikovic, I., Szargel, R., Hawash, A., Labay, V., Hatib, I., Cohen, N. et al. A novel mutation in the chloride channel gene, CLCNKB, as a cause of Gitelman and Bartter syndromes. Kidney Int. 63, 24–32 (2003).
Konrad, M., Vollmer, M., Lemmink, H. H., van den Heuvel, L. P., Jeck, N., Vargas-Poussou, R. et al. Mutations in the chloride channel gene CLCNKB as a cause of classic Bartter syndrome. J. Am. Soc. Nephrol. 11, 1449–1459 (2000).
Guay-Woodford, L. M. Bartter syndrome: unraveling the pathophysiologic enigma. Am. J. Med. 105, 151–161 (1998).
Peters, M., Jeck, N., Reinalter, S., Leonhardt, A., Tönshoff, B., Klaus, G. Gü. et al. Clinical presentation of genetically defined patients with hypokalemic salt-losing tubulopathies. Am. J. Med. 112, 183–190 (2002).
Coto, E., Rodriguez, J., Jeck, N., Alvarez, V., Stone, R., Loris, C. et al. A new mutation (intron 9 +1 G>T) in the SLC12A3 gene is linked to Gitelman syndrome in Gypsies. Kidney Int. 65, 25–29 (2004).
Rothberg, J. M., Hinz, W., Rearick, T. M., Schultz, J., Mileski, W., Davey, M. et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature 475, 348–352 (2011).
Loman, N. J., Misra, R. V., Dallman, T. J., Constantinidou, C., Gharbia, S. E., Wain, J. et al. Performance comparison of benchtop high-throughput sequencing platforms. Nat. Biotechnol. 30, 434–439 (2012).
Harakalova, M., Nijman, I. J., Medic, J., Mokry, M., Renkens, I., Blankensteijn, J. D. et al. Genomic DNA pooling strategy for next-generation sequencing-based rare variant discovery in abdominal aortic aneurysm regions of interest-challenges and limitations. J. Cardiovasc. Transl. Res. 4, 271–280 (2011).
Elliott, A. M., Radecki, J., Moghis, B., Li, X. & Kammesheidt, A. Rapid detection of the ACMG/ACOG-recommended 23 CFTR disease-causing mutations using ion torrent semiconductor sequencing. J. Biomol. Tech. 23, 24–30 (2012).
Costa, J. L., Sousa, S., Justino, A., Kay, T., Fernandes, S., Cirnes, L. et al. Nonoptical massive parallel DNA sequencing of BRCA1 and BRCA2 genes in a diagnostic setting. Hum. Mutat. 34, 629–635 (2013).
Gómez, J., Reguero, J. R., Morís, C., Alvarez, V. & Coto, E. Non optical semi-conductor next generation sequencing of the main cardiac qt-interval duration genes in pooled DNA samples. J. Cardiovasc. Transl. Res. 7, 133–137 (2013).
Tsogalis, G. J., Peterson, J. D., de Abreu, F. B., Tunkey, C. D., Gallagher, T. L., Strausbaugh, L. D. et al. Routine use of the Ion Torrent AmpliSeq Cancer Hotspot Panel for identification of clinically actionable somatic mutations. Clin. Chem. Lab. Med. 52, 707–714 (2013).
Otto, E. A., Ramaswami, G., Janssen, S., Chaki, M., Allen, S. J., Zhou, W. et al. Mutation analysis of 18 nephronophthisis associated ciliopathy disease genes using a DNA pooling and next generation sequencing strategy. J. Med. Genet. 48, 105–116 (2011).
Li, X., Buckton, A. J., Wilkinson, S. L., John, S., Walsh, R., Novotny, T. et al. Towards clinical molecular diagnosis of inherited cardiac conditions: a comparison of bench-top genome DNA sequencers. PLoS ONE 8, e67744 (2013).
Mitsui, J., Fukuda, Y., Azuma, K., Tozaki, H., Ishiura, H., Takahashi, Y. et al. Multiplexed resequencing analysis to identify rare variants in pooled DNA with barcode indexing using next-generation sequencer. J. Hum. Genet. 55, 448–455 (2010).
Missirian, V., Comai, L. & Filkov, V. Statistical mutation calling from sequenced overlapping DNA pools in TILLING experiments. BMC Bioinformatics 12, e287 (2011).
Acknowledgements
We thank Belén Alonso, Sara Iglesias, Ana I. Corao and Marta Díaz for technical support. This work was supported by a grant from Instituto de Salud Carlos III-Fondo Europeo de Desarrollo Regional (FIS-12/00287; FIS-RD12/0021/0012; FIS PI11/01672). BT is a FICYT-Severo Ochoa predoctoral fellow.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Tavira, B., Gómez, J., Santos, F. et al. A labor- and cost-effective non-optical semiconductor (Ion Torrent) next-generation sequencing of the SLC12A3 and CLCNKA/B genes in Gitelman’s syndrome patients. J Hum Genet 59, 376–380 (2014). https://doi.org/10.1038/jhg.2014.37
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2014.37
This article is cited by
-
Allele-specific RT-PCR for the rapid detection of recurrent SLC12A3 mutations for Gitelman syndrome
npj Genomic Medicine (2021)
-
Digenetic inheritance of SLC12A3 and CLCNKB genes in a Chinese girl with Gitelman syndrome
BMC Pediatrics (2019)
-
A Next-Generation Sequencing of the NOTCH3 and HTRA1 Genes in CADASIL Patients
Journal of Molecular Neuroscience (2015)
