
Abstract
When compared with single gene functional analysis, gene set analysis (GSA) can extract more information from gene expression profiles. Currently, several gene set methods have been proposed, but most of the methods cannot detect gene sets with a large number of minor-effect genes. Here, we propose a novel distance-based gene set analysis method. The distance between two groups of genes with different phenotypes based on gene expression should be larger if a certain gene set is significantly associated with the given phenotype. We calculated the distance between two groups with different phenotypes, estimated the significant P-values using two permutation methods and performed multiple hypothesis testing adjustments. This method was performed on one simulated data set and three real data sets. After a comparison and literature verification, we determined that the gene resampling-based permutation method is more suitable for GSA, and the centroid statistical and average linkage statistical distance methods are efficient, especially in detecting gene sets containing more minor-effect genes. We believe that this distance-based method will assist us in finding functional gene sets that are significantly related to a complex trait. Additionally, we have prepared a simple and publically available Perl and R package (http://bioinfo.hrbmu.edu.cn/dbgsa or http://cran.r-project.org/web/packages/DBGSA/).
Similar content being viewed by others
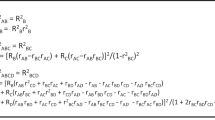
Introduction
With the development of biochip technology and microarrays that contain tens of thousands of genes, we can determine functional gene sets that are related to a phenotype using a gene function enrichment analysis method. There are two primary types of gene function enrichment analysis methods, individual gene analysis (IGA) and gene set analysis (GSA).1, 2, 3 IGA identifies differentially expressed genes through a variety of methods and tests the difference of the proportion of differentially expressed genes between all genes and a given gene set.4, 5 GSA directly calculates gene subset scores using various statistical methods and calculates the significance level.6 The IGA method requires an initial calculation of differentially expressed genes that is influenced by the statistical methods and their thresholds. Since the emergence of gene set enrichment analysis (GSEA), an increasing number of GSA approaches based on various statistical methods have been rapidly developed, such as GSEA,7, 8 globaltest,9 SAM-GS,10 GlobalANCOVA,11 ADGO12, 13 and Bayesian network-based pathway analysis.14
Tian et al.15 classified two types of null hypotheses that test whether a gene set displays a coordinated association with a phenotype of interest. The first type hypothesizes that the genes in a gene set have the same pattern of associations with the given phenotype when compared with the remaining genes (i.e., Q1). The second type hypothesizes that the gene set does not contain any genes that are associated with the given phenotype (i.e., Q2). Geoman and Buhlmann16 termed competitive and self-contained methods based on Q1 and Q2, respectively. These methods have been widely used in previous studies. The genes can be divided into three categories, disease-related genes, minor-effect genes and disease-unrelated genes. The disease-related genes are significantly differentially expressed. The minor-effect genes individually exhibit marginal differential expression but may have a significant combined effect on the phenotypic outcome, such as disease.17 The disease-unrelated genes exhibit no effect on the given phenotype. Usually, there are numerous genes that have a relatively minor-effect in complex diseases.18, 19 Therefore, it is critical to consider the minor-effect genes. Most of the gene function enrichment analysis methods can detect gene sets containing many disease-related genes well. However, they cannot detect the gene sets containing many minor-effect genes.
Here, we propose a novel distance-based gene set enrichment analysis method. We use the original gene expression data, not a summary statistic, in the analysis; therefore, this method utilizes each level of the gene expression data and is better able to detect functional sets, especially for gene sets containing more minor-effect genes. By focusing on gene expression, the distance between two groups with different phenotypes should be larger if a certain gene functional set is significantly associated with a particular phenotype. We use four point-to-point distance measures and two set-to-set distance measures to calculate the distance between two groups with different phenotypes, the case and control groups, by focusing on the gene expression profile of a given gene set. Next, we estimate a significant P-value for this gene set using permutation methods based on the two hypotheses above (Q1 and Q2) and perform multiple hypothesis testing adjustments using the false-discovery rate (FDR). We perform these methods on one simulated data set and three gene expression data sets and compare them with other methods.
In a parallel side study, we first transformed gene expression data to pathway activities using pathway-based microarray analysis methods, including the condition-responsive genes based (CORG-based),20 negatively correlated feature sets with ideal markers (NCFS-i) and negatively correlated feature sets using the CORG-based method (NCFS-c) methods,21, 22 and then analyzed them with methods such as GSA or disease classification. In this manuscript, we present an improvement of these three methods for the detection of disease-related pathways.
Materials and methods
Data
Gene expression profiles
To analyze whether this method is feasible and effective, we used one simulated data set and three real data sets.
Simulated data set. We simulated a data set of 50 cases and 50 controls that included 10 000 genes for 100 samples. There was no exact proportion of the disease-related genes, the minor-effect genes and the disease-unrelated genes, but we thought there should be more minor-effect genes than significant disease-related genes. Therefore, we used proportions of 10% (1000 significant disease-related genes), 30% (3000 minor-effect genes) and 60% (6000 disease-unrelated genes) in this simulation. Reuben Thomas et al.23 reported that a priori assumption of any of the considered univariate theoretical probability distributions across all probe sets was not valid. There was no assumption of probability distribution in our proposed methods. However, we needed to build a data set that included significantly differentially expressed genes and non-differentially expressed genes. The common and effective way to do this was to assume that the gene expression followed a normal distribution because we could easily control whether the genes were differentially expressed. The normal distribution was only used for the simulation purpose and we did not need any assumption of probability distribution in the proposed methods. We generated the disease-related genes as follows: 500 were downregulated genes that followed the normal distributions N (0,1) in the case group and N (1,1) in the control group. A total of 500 of the genes were upregulated and followed normal distributions N (1,1) in the case group and N (0,1) in the control group. All of the genes were significantly differentially expressed using a t-test with a significance level of 0.001. We generated the minor-effect genes as follows: 1500 of the genes followed normal distributions N (0,1) in the case group and N (0.5,1) in the control group. Fifteen hundred of the genes followed normal distributions N (0.5,1) in the case group and N (0,1) in the control group. About half of the genes were significantly differentially expressed using a t-test with a significance level of 0.001. So we considered them minor-effect genes. We generated the disease-unrelated genes as follows: 6000 genes followed a normal distribution N (0,1) in the case and control groups. None of the genes was significantly differentially expressed using a t-test with a significance level of 0.001.
Real data sets. Alzheimer’s disease (AD) is a common neurodegenerative disease that severely affects the quality of life of the elderly. We selected an AD gene expression data set from Gene Expression Omnibus (GEO, GSE15222).24 This data set included 24 350 probes, 363 samples, 187 controls and 176 cases. First, we performed pretreatments, including using the mean value of gene expression when multiple probes corresponded to one gene and removing missing data lines and outliers. We suggest performing these pretreatments before the GSA. Finally, we obtained expression values for 17 007 genes.
Non-small cell lung cancer (NSCLC) is a broad term for lung cancers that are not of the small-cell type. The three most common subtypes of NSCLC include adenocarcinoma (AC), squamous cell carcinoma (SCC) and large-cell carcinoma (LCC). We obtained two gene expression profiles of high-grade human NSCLC specimens. One data set, NSCLC I, included 58 samples (40 AC samples and 18 SCC samples, GSE10245).25 After pretreating the data, we obtained expression values for 19 801 genes. The other data set, NSCLC II, included 28 samples (9 AC samples and 19 SCC samples, GSE27388).26 After pretreating the data, we obtained expression values for 18 302 genes. We performed a Gene ontology (GO) functional set analysis for these data.
Gene sets. For the simulated data set, we constructed 1600 gene sets with different set sizes and proportions of different genes. We constructed 200 gene sets, 50 presumed disease-related gene sets containing more disease-related genes, 50 presumed disease-related gene sets containing more minor-effect and 100 presumed disease-unrelated gene sets, for set sizes of 10, 20, 30, 40, 50, 100, 150 and 200 genes. The detailed instructions are shown in Table 1.
For the real data sets, the gene sets were derived from the Molecular Signatures Database (MSigDB).8 There are 6769 gene sets in MSigDB version 3.0 that are divided into five major collections. GO 27 is the most popular and widely used biomedical ontology. It is the de facto standard for effective functional annotation and enrichment analysis of high-throughput gene expression data sets. We used the GO gene sets, which are part of MSigDB v3.0 in this study. Because gene sets with too many or too few genes are uninformative, only 1401 GO gene sets with 10 to 500 genes were used.
Methods
Distance-based methods
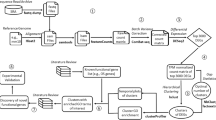
First, we calculate the distance between the case and control groups by focusing on the gene expression for a given gene set. Next, we estimate the significant P-values for this gene set. And then we perform multiple hypothesis testing adjustments by FDR. The flow chart for this method is shown in Figure 1. The three key steps of the distance-based gene set analysis (DBGSA) method are described below.

Flow chart of distance-based gene set analysis (DBGSA).
Step 1: Calculate the distance between two groups with different phenotypes in a given gene set
First, we combine the gene expression profile and a given gene set from MSigDB to obtain a gene expression subset. Suppose that there are t individuals in the gene expression profile, which include t1 individuals from the case group and t2 individuals from the control group, and there are n genes in a gene set, which includes m genes in the gene expression profile. Here, we consider one person’s gene expression values as a point in m dimensional space. Therefore, we can obtain t1 and t2 points in the case and control groups, respectively. The distance measures between the points and between the sets are defined below. We denote dij0 to be the distance between objects i and j and xik to be the kth gene expression value of person i.
The definitions of the distance between points are as follows.
Euclidean distance (-euc)

Statistical distance (-stat)

where skk is the sample variance of variable xik. The statistical distance is considered a weighted Euclidean distance using  as the weight.
as the weight.
Manhattan distance (-man)

Chebyshev distance (-max)  The definition of the distance between the case and control sets is as follows.
The definition of the distance between the case and control sets is as follows.
Average linkage method (avelink-)
The average linkage method specifies that the distance between two sets is computed as the average distance between the objects from a set (case set Gcase) and the objects from another set (control set Gcontrol). The averaging is performed over all pairs (i, j) of objects, where i is an object from case set Gcase and j is an object from control set Gcontrol. This can be mathematically described as

where t1 and t2 are the sample numbers of set Gcase and Gcontrol and dij is the distance between i from Gcase and j from Gcontrol.
Centroid method (cent-)
The centroid method, which specifies the distance between two sets, is computed as the distance between the centroids of two sets. Mathematically, this method can be described as
 where
where  and
and  are the centroid of sets Gcase and Gcontrol.
are the centroid of sets Gcase and Gcontrol.
Each time, we select a definition of the distance measures between the points and between the sets. Therefore, we obtain eight different combinations of distance measures. We use the abbreviation of each combination of distance measures in the following analyses, such as avelink-euc for the average linkage Euclidean distance method. We denote the distance between the case and control sets as D0.
Step 2: Estimate the significant level of the gene sets
We use permutation to estimate the significance level of the gene sets and perform two types of permutations according to Q1 and Q2.
Gene resampling-based permutation
There are two main methods in resampling theory, using subsets of available data (e.g., jackknifing) and drawing randomly with replacement from a set of data points (e.g., bootstrapping).28, 29, 30 Because the presence of two or more of the same genes in one gene set is unlikely, we use the resampling method without replacement.
We randomly resample mi genes from the gene expression profile and obtain a subset of gene expression profiles with s individuals and mi genes. Next, we calculate the distances between the two groups with different phenotypes according to the method described in step 1. This procedure is repeated nper times to obtain nper distances randomly, which are denoted as 
To screen out functional sets that are significantly associated with disease, we calculate P-values by a rank-based method as follows.

Label swapping-based permutation
For a specific gene expression profile set, we swap the label of each individual by controlling the same total number of cases and controls so as to obtain a new gene expression profile set. Next, we calculate the distance between the two groups with different phenotypes according to the method that was described in step 1. Similar to gene resampling, we repeat this procedure nper times to randomly obtain nper distances, and we denote them as  We also calculate the P-value as follows:
We also calculate the P-value as follows:

Step 3: Adjusted for multiple hypothesis testing
The estimated significance level should be adjusted to account for multiple hypothesis testing when thousands of gene sets are tested. The calculation of FDR has been shown to be an effective method. We use fdrtool31 to estimate the tail area-based FDR (Fdr) and density-based local FDR in this study.
Using these three steps, we can determine whether a gene set is significantly related to a trait.
Gene set enrichment analysis
GSEA is a widely used gene set analysis method. We use a gene resampling-based GSEA method for comparison in this study.
CORG-based method, NCFS-i method and NCFS-c method
CORG-based, NCFS-i and NCFS-c methods are efficient pathway-based microarray analysis methods.20, 21, 22 We present an improvement based on these three methods for the detection of disease-related pathways. First, we use these original methods to select a subset for each gene set. Next, we use five-fold cross validation to calculate the accuracy of the subset in disease classification. Finally, we classify the top gene sets with high accuracy as the disease-related gene sets. Instead of P-values, we choose the significance threshold to be a classification of accuracy.
Precision, recall and the F-measure (F1)
We use precision, recall and the F-measure (F1) to evaluate different methods for detecting disease-related gene sets. These terms are defined as follows:



The terms TP, TN, FP and FN represent true positives, true negatives, false positives and false negatives, respectively.
Overlap coefficient
While measuring the similarity of two sets with large differences in set sizes, we should pay more attention to the smaller set. The overlap coefficient32, 33 is a proper similarity measure in this situation that computes the overlap between two sets and is defined as follows:

where X and Y are two sets and |X| indicates the set size of X.
Results and discussion
In this section, the proposed method was compared with other methods. First, we evaluated the label swapping-based permutation method and found that it was improper. Next, we evaluated the gene resampling-based permutation method in the simulated data set and found that the avelink-stat and cent-stat methods were appropriate for GSA. Additionally, we compared these methods with GSEA, the CORG-based method, the NCFS-i method and the NCFS-c method using various evaluation measures in the simulated data set and the real data sets. The details are shown in Figure 2.

Flow chart of the comparison and evaluation.
Results from the label swapping-based permutation
For the AD data, we found that most of the functional sets were significantly associated with AD by our label swapping-based permutation method; the results are shown in Table 2.
To understand these results, we performed a simulation experiment. We selected different numbers of disease-unrelated genes and disease-related genes from the simulated data set to form gene sets (gene set sizes ranging from 10 to 200 genes in 5 gradients). And then we calculated P-values using the avelink-euc method by performing 1000 permutations. We repeated this procedure 10 times for each case and observed the number of significant results. These results are shown in Table 3.
If there are more disease-related genes in a gene set with a given set size, then there is a higher probability that the gene set detected is disease-related. In the simulated data, the proportion of the disease-related genes is 10%. However, the results indicate that although there were only two disease-related genes in a tested gene set containing 200 genes (the proportion of the disease-related genes is just 1%), we could still detect the gene set significantly related to disease.
We also evaluated this method in the real AD data set. We randomly selected several genes (gene set sizes ranging from 10 to 200 genes in 5 gradients) to form a putative functional category, calculated P-values using the avelink-euc distance method by performing 1000 permutations, and repeated the procedure 100 times for each case. Next, we calculated the number of significant results using a significance level of 0.001. These results are shown in Table 4.
Based on the results, even the random selection of genes from the AD data set would produce significant results with a probability of >85%, indicating that many false disease-related gene sets (false positives) may be found. The simulated and real data indicated that this label swapping-based permutation method was too sensitive and may result in high false-positive rates in the detection of significant disease-related functional gene sets.
Results from the gene resampling-based permutation
Comparison of the simulated data set
We performed gene resampling-based GSEA, the CORG-based method, the NCFS-i method, the NCFS-c method and our proposed gene resampling-based permutation method on the simulated data set. We selected P<0.05 as the significance threshold for the gene resampling-based GSEA and our gene resampling-based permutation method. Because there is no empirical threshold for the CORG-based, NCFS-i and NCFS-c methods, we selected a 5-fold accuracy >0.9, >0.85 and >0.8 as thresholds. We counted the number of statistically significant disease-related gene sets using these methods and calculated the precision, recall and F1 scores. These results are shown in Table 5.
We aimed to select the most efficient of our eight proposed methods. From the results, we found that the avelink-stat method performed best out of the average linkage methods, and the cent-euc, cent-man and cent-stat methods performed similarly out of the centroid methods. Because the variance in our simulated data set was set to 1 for all the genes, we replaced 10 of the minor-effect genes with larger variances to test the robustness of our methods for unnormalized data. In these 10 genes, the expression followed a normal distribution N (10,1000) in the case group and a normal distribution N (0,1000) in the control group. We constructed 100 disease-unrelated gene sets within 100 genes as follows. In each gene set, we randomly selected 10 disease-related genes (10%), 1 minor-effect gene with a variance of 1000 and 29 minor-effect genes with a variance of 1 (30%), and 60 disease-unrelated genes (60%). We suggested that these 100 gene sets were unrelated to disease because the proportions of different gene types were the same as the total data set. We performed the proposed gene resampling-based methods on this data set, selected P<0.05 as the significance threshold and counted the number of significant disease-related gene sets. The results indicate that only the avelink-stat and cent-stat methods are robust for the dimensions of the gene expression values; therefore, we used these two methods in the following analyses. The results are shown in Table 5.
Next, we compared the avelink-stat, cent-stat, CORG-based, NCFS-i and NCFS-c methods and gene resampling-based GSEA. The gene set size influenced the results, such that more significant results were obtained with larger gene set sizes. In the CORG-based, NCFS-i and NCFS-c methods, the influences of the threshold in the gene sets with different gene set sizes differed. When the gene set size was small, more truly significant disease-related gene sets were obtained using a lower acc threshold (acc>0.8). But when the gene set size was large, too many false significant disease-related gene sets were obtained using a lower acc threshold (97, 99 and 99 false positives in 100 negative gene sets when the gene set size is 200 using acc >0.8 as the threshold). In the gene set analysis, we wanted more accurate results with a lower FDR. Therefore, we selected the higher acc threshold (acc>0.9) for these three methods. For precision, the avelink-stat and cent-stat methods resulted in higher values than GSEA when the gene set sizes were small (<50); the avelink-stat and cent-stat methods resulted in higher values than the CORG-based, NCFS-i and NCFS-c methods when the gene set sizes were large (>100) because the CORG-based, NCFS-i and NCFS-c methods resulted in more false-negative gene sets than the proposed two methods (e.g., 30, 48 and 50 false negatives in these three methods vs no false negatives in our two methods when the gene set size is 200). The overall precisions (1600 gene sets) of these six methods were 1, 1, 0.86, 0.92, 0.85 and 0.86, respectively. For recall, the avelink-stat and cent-stat methods resulted in higher values than the four other methods for all gene set sizes. The overall recall values for these six methods were 0.83, 0.85, 0.39, 0.37, 0.56 and 0.56, respectively, because our methods resulted in more true-positive gene sets, especially those gene sets containing more minor-effect genes (e.g., 42 and 43 true positive gene sets containing more minor-effect genes in our methods vs 9, 4, 10 and 18 in the other 4 methods when the gene set size was 50). The overall F1 scores of these six methods were 0.91, 0.92, 0.54, 0.53, 0.68 and 0.68, respectively, which demonstrates that our methods were best. We also compared these methods in negative gene sets with unnormalized data. The significant false-positive gene sets from the 100 negative gene sets were 0, 0, 3, 22, 64 and 67 using these six methods, respectively. Therefore, our two methods and gene resampling-based GSEA were robust for the unnormalized data. Taking into consideration the overall precision, recall and F1 score of the 1600 gene sets, we believe that both the avelink-stat method (1, 0.83 and 0.91, respectively) and the cent-stat method (1, 0.85 and 0.92, respectively) are better suited for GSA and perform better than other methods, especially in detecting disease-related gene sets containing more minor-effect genes and in their robustness for handling unnormalized data. We obtained a significantly higher overall recall of the 1600 gene sets using the cent-stat method (0.85) compared with the avelink-stat method (0.83) or other methods (GSEA, 0.39; the CORG-based, 0.37; NCFS-i, 0.56; and NCFS-c, 0.5625). Therefore, the cent-stat method was more sensitive than the avelink-stat and other methods. Thus, we performed these two methods on three real data sets.
A comparison of the avelink-stat method and the cent-stat method in real data sets
We calculated the average distance between each point from two gene sets for the average linkage distance method. Next, we determined the centroid of each gene set and calculated the distances between the two centroids for the centroid distance method. To further compare the detection ability of the two methods, we selected a Fdr<0.05 as the significance threshold of association between the gene sets and traits in the real data sets. For the AD data, we obtained 10 statistically significant functional sets using the avelink-stat method and 116 sets using the cent-stat method; 8 of these sets were included in both of the methods. For the NSCLC I data, we obtained 91 statistically significant functional sets using the avelink-stat method and 129 sets using the cent-stat method; 68 of these sets were included in both of the methods. For the NSCLC II data, we obtained 321 statistically significant functional sets using the avelink-stat method and 495 sets using the cent-stat method; 304 of these sets were included in both of the methods. In these examples, we obtained more significant functional sets using the cent-stat method than the avelink-stat method. Detailed results are shown in Table 6.
A comparison of the distance-based method and other methods in real data sets
In addition, we performed gene resampling-based GSEA, the CORG-based method, the NCFS-i method and the NCFS-c method using these three data sets. We calculated the correlation coefficients of the GSEA, avelink-stat and cent-stat methods. The results are shown in Table 7. The correlation between the cent-stat method and the avelink-stat method was greater than the correlations between the cent-stat method and GSEA and between the avelink-stat method and GSEA. The detailed computational results for the AD data are shown in Supplementary Table 1.
We selected a Fdr <0.05 as the significance threshold for the GSEA analysis. Moreover, we selected an acc>0.9 as the significance threshold for the CORG-based, NCFS-i and NCFS-c methods based on the analysis of the threshold choice in the simulated data.
For the AD data, we obtained 42 statistically significant functional gene sets using GSEA. One of these sets was shared between GSEA and the avelink-stat method, and 23 sets were shared between GSEA and the cent-stat method; one set was shared by all three methods. The results are shown in Table 6. More functional sets were obtained using the cent-stat method than GSEA, which included more detailed portions of the functional sets. For example, we obtained ‘regulation of transcription, DNA dependent’ using GSEA. We obtained some additional depth nodes by the cent-stat method, such as ‘regulation of transcription factor activity’ and ‘negative regulation of transcription DNA dependent’. We did not obtain any statistically significant functional gene sets using the CORG-based, NCFS-i and NCFS-c methods. Even when we set the acc >0.8 as the significance threshold, we could not obtain any statistically significant functional gene set using these methods.
For the NSCLC I data, we obtained 63 statistically significant functional sets using GSEA. A total of 31 of these sets were shared between GSEA and the avelink-stat method, 51 were shared between GSEA and the cent-stat method, and 30 were shared by all three methods. For the NSCLC II data, we obtained 399 statistically significant functional sets using GSEA. Overall, 189 of these sets were shared between GSEA and the avelink-stat method, 278 were shared between GSEA and the cent-stat method, and 189 were shared by all three methods. These results are shown in Table 6 and show that more significant disease-related gene sets are obtained using the cent-stat method than gene resampling-based GSEA.
For the NSCLC I data, we obtained 194, 181 and 179 statistically significant functional sets using the CORG-based, NCFS-i and NCFS-c methods, respectively. A total of 63 of these sets were shared by the three methods. For the NSCLC II data, we obtained 393, 438 and 425 statistically significant functional sets using the CORG-based, NCFS-i and NCFS-c methods, respectively. Overall, 268 of these sets were shared by the three methods. These results are shown in Table 6. We obtained as many significant gene sets using the CORG-based, NCFS-i and NCFS-c methods as using the distance-based methods.
Literature verification
We performed a literature verification to determine whether the significant functional gene sets obtained from different methods were associated with the trait. For the AD data, we acquired 10 significant functional sets from the avelink-stat method. We found that 9 out of the 10 gene sets were already recognized in a large number of AD literature sources. Additionally, we searched for the top 10 significant GO terms obtained by GSEA, CORG-based method, NCFS-i and NCFS-c, respectively.34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62 We confirmed that eight of them from GSEA, seven of them from CORG-based method, eight of them from NCFS-i and seven of them from NCFS-c were verified in the literature. There was a linear relation between the recall and the total number of actual disease-related gene sets found by the method in the same data. So the recall of the avelink-stat method was higher than others. There was only one overlapping GO term between the avelink-stat method and GSEA, and no overlapping GO term between the avelink-stat method and CORG-based method, NCFS-i and NCFS-c. However, there were seven overlapping GO terms between NCFS-i and NCFS-c. The results are shown in Table 8, the functional sets verified in the literature are marked with the letter ‘a’, and the overlapping gene sets are shown in bold.
Moreover, we performed literature verification for the significant functional gene sets obtained from the cent-stat method for AD. We verified 68 out of 116 (58.6%) gene sets in the literature. From the 23 sets in common with GSEA, 13 (56.5%) were verified; from the 93 sets that were different from GSEA, 55 (59.1%) were verified. From GSEA, 22 out of 42 gene sets were verified in the literature. The recall of the cent-stat method was significantly higher than the avelink-stat method and GSEA. In other words, the cent-stat method was more sensitive than the avelink-stat method and GSEA. We obtained a significantly higher precision value using our proposed methods (0.90 and 0.59) than GSEA (0.52). Particularly, we obtained a precision of 1 using a combination of the avelink-stat method and the cent-stat method. Additional functional gene sets that were not verified in the literature may be associated with AD. For example, prior studies found that ‘actin binding’,63 ‘actin cytoskeleton organization and biogenesis’,64 ‘actin filament binding’63 and ‘actin filament organization’65 were related to AD, and we identified two additional functional gene sets, ‘actin filament based process’ and ‘actin filament bundle formation’. From the relationships between these sets, we believed that the two newly discovered functional sets were related to AD. This example demonstrated the effectiveness of these methods. The detailed results of this analysis are shown in Supplementary Table 2.
Using the NSCLC I data, Ruprecht Kuner25 found 23 significant functional GO terms using GOstat66 at a significance level of P<0.0001. From these 23 GO terms, 10, 11, 7, 5, 8 and 9 terms were detected by the avelink-stat method, the cent-stat method, GSEA, the CORG-based method, the NCFS-i method and the NCFS-c method, respectively. Our proposed methods yielded more verified disease-related gene sets than other methods, even if we obtained more significant gene sets using the CORG-based, NCFS-i and NCFS-c methods. We obtained a significantly higher precision value using a combination of the avelink-stat method and the cent-stat method (0.15) than other methods (<0.11). In addition, we obtained significantly higher recall values using our proposed methods (>0.43) than other methods (<0.39). Specifically, the term ‘cell junctions’ that was previously described25 was detected by both of our methods but was not found using GSEA, the CORG-based method or the NCFS-i method. These results are shown in Table 6.
Computational complexity and repeatability
In the gene resampling-based permutation methods, gene resampling is the most time-consuming step. Suppose that we randomly resample mi genes from a gene expression profile including m genes, calculate the distances between the two groups with different phenotypes and repeat this procedure nper times. The computational complexity of these steps is approximately O (Npermmi). Even when nper and m are large, the computational time is still acceptable.
The repeatability of the method is very important when detecting disease-related gene sets. We used the overlap coefficient and correlation coefficient between the two NSCLC data sets to describe the repeatability. These results are shown in Table 6 and Table 7b and c. We obtained a greater overlap coefficient while considering a combination of the avelink-stat and cent-stat methods (85.3%) compared with GSEA (77.8%) and the three other methods (a maximum of 64.4%). The Pearson’s and Spearman correlation coefficients between the two data sets using the avelink-stat method (0.4290 and 0.4421, respectively) and the cent-stat method (0.4365 and 0.4785, respectively) were significantly larger than that with GSEA (0.0485 and 0.1326, respectively) and the CORG-based, NCFS-i and NCFS-c methods (Pearson’s correlation coefficients of 0.4316, 0.4028 and 0.3844, and Spearman correlation coefficients of 0.4015, 0.3957 and 0.3731, respectively). These results confirmed that our proposed methods had better repeatability than other methods.
Conclusion
Compared with the IGA methods, we do not need to set a threshold for expression difference to classify genes between the case and control samples. In addition, we use the original gene data and not a summary statistic during analysis, whereas most GSA methods use summary statistics, such as the rank statistic used by GSEA. Therefore, this method fully utilizes each level of the gene expression data and is better able to detect functional sets, especially for gene sets containing more minor-effect genes. By analyzing simulated and real data, we determined that the label swapping-based permutation method is too sensitive and may result in high false-positives during the detection of significant disease-related functional gene sets; by evaluating precision, recall and the F1 scores in the simulated data, we believe that the gene resampling-based permutation method is more suitable for gene set analyses. For the gene resampling-based permutation method, we determine that the statistical distance method is robust for the dimensions of the gene expression values.
Compared with GSEA and the CORG-based, NCFS-i and NCFS-c methods in the simulation experiment, we find that both the cent-stat and avelink-stat methods perform best, especially in detecting the disease-related gene sets containing more minor-effect genes.
When using the real data sets, we find that the cent-stat method is more sensitive than the avelink-stat method and other methods. The precision obtained from a combination of the avelink-stat and cent-stat methods is higher than the precision of other methods. Through validation using duplicate data sets, we determine that the repeatability of a combination of these two methods is better than other methods. Therefore, we recommend the use of the cent-stat method for the identification of more functional gene sets and a combination of these two methods for the more accurate identification of disease-related functional gene sets.
In this study, we perform distance-based gene set analysis with strong feasibility and effectiveness using GO as examples. In addition, we can analyze other functional sets, such as KEGG pathways and motif gene sets. We have prepared a simple and publically available Perl and R package for the centroid statistical distance method and the average linkage statistical distance method (http://bioinfo.hrbmu.edu.cn/dbgsa or http://cran.r-project.org/web/packages/DBGSA/).
References
Nam, D. & Kim, S. Y. Gene-set approach for expression pattern analysis. Brief. Bioinform. 9, 189–197 (2008).
Emmert-Streib, F. & Glazko, G. V. Pathway analysis of expression data: deciphering functional building blocks of complex diseases. PLoS Comput. Biol. 7, e1002053 (2011).
Hung, J. H., Yang, T. H., Hu, Z., Weng, Z. & Delisi, C. Gene set enrichment analysis: performance evaluation and usage guidelines. Brief Bioinform 13, 281–291 (2012).
Khatri, P. & Draghici, S. Ontological analysis of gene expression data: current tools, limitations, and open problems. Bioinformatics 21, 3587–3595 (2005).
Rivals, I., Personnaz, L., Taing, L. & Potier, M. C. Enrichment or depletion of a GO category within a class of genes: which test? Bioinformatics 23, 401–407 (2007).
Dopazo, J. Functional interpretation of microarray experiments. OMICS 10, 398–410 (2006).
Mootha, V. K., Lindgren, C. M., Eriksson, K. F., Subramanian, A., Sihag, S., Lehar, J. et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273 (2003).
Subramanian, A., Tamayo, P., Mootha, V. K., Mukherjee, S., Ebert, B. L., Gillette, M. A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Goeman, J. J., van de Geer, S. A., de Kort, F. & van Houwelingen, H. C. A global test for groups of genes: testing association with a clinical outcome. Bioinformatics 20, 93–99 (2004).
Dinu, I., Potter, J. D., Mueller, T., Liu, Q., Adewale, A. J., Jhangri, G. S. et al. Improving gene set analysis of microarray data by SAM-GS. BMC Bioinform. 8, 242 (2007).
Hummel, M., Meister, R. & Mansmann, U. GlobalANCOVA: exploration and assessment of gene group effects. Bioinformatics 24, 78–85 (2008).
Nam, D., Kim, S. B., Kim, S. K., Yang, S., Kim, S. Y. & Chu, I. S. ADGO: analysis of differentially expressed gene sets using composite GO annotation. Bioinformatics 22, 2249–2253 (2006).
Chi, S. M., Kim, J., Kim, S. Y. & Nam, D. ADGO 2.0: interpreting microarray data and list of genes using composite annotations. Nucleic Acids Res 39, W302–W306 (2011).
Isci, S., Ozturk, C., Jones, J. & Otu, H. H. Pathway analysis of high-throughput biological data within a Bayesian network framework. Bioinformatics 27, 1667–1674 (2011).
Tian, L., Greenberg, S. A., Kong, S. W., Altschuler, J., Kohane, I. S. & Park, P. J. Discovering statistically significant pathways in expression profiling studies. Proc. Natl Acad. Sci. USA 102, 13544–13549 (2005).
Goeman, J. J. & Buhlmann, P. Analyzing gene expression data in terms of gene sets: methodological issues. Bioinformatics 23, 980–987 (2007).
Ye, C. & Eskin, E. Discovering tightly regulated and differentially expressed gene sets in whole genome expression data. Bioinformatics 23, e84–e90 (2007).
Thomson, G. An overview of the genetic analysis of complex diseases, with reference to type 1 diabetes. Best practice & research Clinical endocrinology & metabolism. Best Pract. Res. Clin. Endocrinol. Metab 15, 265–277 (2001) [Research Support, US Govt PHS Review].
Scott, W. K., Pericak-Vance, M. A. & Haines, J. L. Genetic analysis of complex diseases. Science 275, 1327–1330 (1997).
Lee, E., Chuang, H. Y., Kim, J. W., Ideker, T. & Lee, D. Inferring pathway activity toward precise disease classification. PLoS Comput. Biol 4 e1000217 (2008).
Sootanan, P., Prom-on, S., Meechai, A. & Chan, J. Pathway-based microarray analysis for robust disease classification. Neural Comput. Appl. 21, 649–660 (2012).
Chan, J. H., Sootanan, P. & Larpeampaisarl, P. Feature selection of pathway markers for microarray-based disease classification using negatively correlated feature sets. 2011 International Joint Conference on Neural Networks (IJCNN 2011) IEEE. p 3293–3299 (2011).
Thomas, R., de la Torre, L., Chang, X. & Mehrotra, S. Validation and characterization of DNA microarray gene expression data distribution and associated moments. BMC Bioinform. 11, 576 (2010).
Webster, J. A., Gibbs, J. R., Clarke, J., Ray, M., Zhang, W., Holmans, P. et al. Genetic control of human brain transcript expression in Alzheimer disease. Am. J. Hum. Genet. 84, 445–458 (2009) [Research Support, N.I.H., Extramural].
Kuner, R., Muley, T., Meister, M., Ruschhaupt, M., Buness, A., Xu, E. C. et al. Global gene expression analysis reveals specific patterns of cell junctions in non-small cell lung cancer subtypes. Lung Cancer 63, 32–38 (2009).
Hall, J. S., Leong, H. S., Armenoult, L. S., Newton, G. E., Valentine, H. R., Irlam, J. J. et al. Exon-array profiling unlocks clinically and biologically relevant gene signatures from formalin-fixed paraffin-embedded tumour samples. Br. J. Cancer 104, 971–981 (2011).
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 (2000) [Research Support, Non-US Gov’t Research Support, US Gov’t, PHS].
Hjorth, J. S. U. Computer intensive statistical methods validation model selection and bootstrap (Chapman and Hall: London, 1994).
Manly, B. F. J. Randomization, bootstrap, and Monte Carlo methods in biology. 3rd ed. (Chapman & Hall/CRC: Boca Raton, Fla.: London, 2007).
Zieffler, A., Harring, J. & Long, J. D. Comparing groups randomization and bootstrap methods using R. (Wiley-Blackwell: Oxford, 2011).
Strimmer, K. Fdrtool a versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics 24, 1461–1462 (2008).
Chapman, S. SimMetrics: Open Source Similarity Measure Library (2007). Available from: http://sourceforge.net/projects/simmetrics/.
Chapman, S. String similarity metrics for information integration (2007). Available from: http://www.dcs.shef.ac.uk/~sam/stringmetrics.html.
Mecocci, P., MacGarvey, U. & Beal, M. F. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann. Neurol. 36, 747–751 (1994).
Cordell, B. beta-Amyloid formation as a potential therapeutic target for Alzheimer's disease. Annu. Rev. Pharmacol. Toxicol. 34, 69–89 (1994).
Lipton, S. A., Gu, Z. & Nakamura, T. Inflammatory mediators leading to protein misfolding and uncompetitive/fast off-rate drug therapy for neurodegenerative disorders. Int. Rev. Neurobiol. 82, 1–27 (2007).
Tabner, B. J., Turnbull, S., El-Agnaf, O. & Allsop, D. Production of reactive oxygen species from aggregating proteins implicated in Alzheimer’s disease, Parkinson’s disease and other neurodegenerative diseases. Curr. Top Med. Chem. 1, 507–517 (2001).
Datta, K., Sinha, S. & Chattopadhyay, P. Reactive oxygen species in health and disease. Natl Med. J. India 13, 304–310 (2000).
Perry, G., Kawai, M., Tabaton, M., Onorato, M., Mulvihill, P., Richey, P. et al. Neuropil threads of Alzheimer’s disease show a marked alteration of the normal cytoskeleton. J. Neurosci. 11, 1748–1755 (1991).
Bamburg, J. R. & Wiggan, O. P. ADF/cofilin and actin dynamics in disease. Trends Cell Biol. 12, 598–605 (2002).
Matus, S., Lisbona, F., Torres, M., Leon, C., Thielen, P. & Hetz, C. The stress rheostat: an interplay between the unfolded protein response (UPR) and autophagy in neurodegeneration. Curr. Mol. Med. 8, 157–172 (2008).
Barnham, K. J., McKinstry, W. J., Multhaup, G., Galatis, D., Morton, C. J., Curtain, C. C. et al. Structure of the Alzheimer’s disease amyloid precursor protein copper binding domain. A regulator of neuronal copper homeostasis. J. Biol. Chem. 278, 17401–17407 (2003).
Lin, C. L., Bristol, L. A., Jin, L., Dykes-Hoberg, M., Crawford, T., Clawson, L. et al. Aberrant RNA processing in a neurodegenerative disease: the cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron 20, 589–602 (1998).
Cook, D. G., Forman, M. S., Sung, J. C., Leight, S., Kolson, D. L., Iwatsubo, T. et al. Alzheimer's A beta (1-42) is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nat. Med. 3, 1021–1023 (1997).
Ebneth, A., Godemann, R., Stamer, K., Illenberger, S., Trinczek, B. & Mandelkow, E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer’s disease. J. Cell. Biol. 143, 777–794 (1998).
Nixon, R. A., Wegiel, J., Kumar, A., Yu, W. H., Peterhoff, C., Cataldo, A. et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 64, 113–122 (2005).
Wallace, D. C. Mitochondrial diseases in man and mouse. Science 283, 1482–1488 (1999).
Geula, C., Greenberg, B. D. & Mesulam, M. M. Cholinesterase activity in the plaques, tangles and angiopathy of Alzheimer’s disease does not emanate from amyloid. Brain Res. 644, 327–330 (1994).
Cutler, R. G., Kelly, J., Storie, K., Pedersen, W. A., Tammara, A., Hatanpaa, K. et al. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl Acad. Sci. USA 101, 2070–2075 (2004).
Stadelmann, C., Deckwerth, T. L., Srinivasan, A., Bancher, C., Bruck, W., Jellinger, K. et al. Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer’s disease. Evidence for apoptotic cell death. Am. J. Pathol. 155, 1459–1466 (1999).
Cassarino, D. S., Swerdlow, R. H., Parks, J. K., Parker, W. D. & Bennett, J. P. Cyclosporin A increases resting mitochondrial membrane potential in SY5Y cells and reverses the depressed mitochondrial membrane potential of Alzheimer's disease cybrids. Biochem. Biophys. Res. Commun. 248, 168–173 (1998).
Toide, K., Okamiya, K., Iwamoto, Y. & Kato, T. Effect of a novel prolyl endopeptidase inhibitor, JTP-4819, on prolyl endopeptidase activity and substance P- and arginine-vasopressin-like immunoreactivity in the brains of aged rats. J. Neurochem. 65, 234–240 (1995).
Connell, C. M., Shaw, B. A., Holmes, S. B., Hudson, M. L., Derry, H. A. & Strecher, V. J. The development of an Alzheimer’s disease channel for the Michigan Interactive Health Kiosk Project. J. Health Commun. 8, 11–22 (2003).
Kaminska, J., Hoffman-Sommer, M. & Plachta, M. The p24 family proteins--regulators of vesicular trafficking. Postepy Biochem. 56, 75–82 (2010).
Ross, B. M., Moszczynska, A., Erlich, J. & Kish, S. J. Phospholipid-metabolizing enzymes in Alzheimer's disease: increased lysophospholipid acyltransferase activity and decreased phospholipase A2 activity. J. Neurochem. 70, 786–793 (1998).
Lee, J. M., Calkins, M. J., Chan, K., Kan, Y. W. & Johnson, J. A. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J. Biol. Chem. 278, 12029–12038 (2003).
Hirai, K., Aliev, G., Nunomura, A., Fujioka, H., Russell, R. L., Atwood, C. S. et al. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 21, 3017–3023 (2001).
David, D. C., Ittner, L. M., Gehrig, P., Nergenau, D., Shepherd, C., Halliday, G. et al. Beta-amyloid treatment of two complementary P301L tau-expressing Alzheimer's disease models reveals similar deregulated cellular processes. Proteomics 6, 6566–6577 (2006).
Perry, T. L., Yong, V. W., Bergeron, C., Hansen, S. & Jones, K. Amino acids, glutathione, and glutathione transferase activity in the brains of patients with Alzheimer’s disease. Ann. Neurol. 21, 331–336 (1987) [Research Support, Non-US Gov’t].
Brinton, R. D. Cellular and molecular mechanisms of estrogen regulation of memory function and neuroprotection against Alzheimer’s disease: recent insights and remaining challenges. Learn Mem. 8, 121–133 (2001).
Baloyannis, S. J. Mitochondrial alterations in Alzheimer’s disease. J. Alzheimers Dis. 9, 119–126 (2006).
Lukiw, W. J. & Bazan, N. G. Strong nuclear factor-kappaB-DNA binding parallels cyclooxygenase-2 gene transcription in aging and in sporadic Alzheimer’s disease superior temporal lobe neocortex. J. Neurosci. Res. 53, 583–592 (1998).
Harigaya, Y., Shoji, M., Shirao, T. & Hirai, S. Disappearance of actin-binding protein, drebrin, from hippocampal synapses in Alzheimer’s disease. J. Neurosci. Res. 43, 87–92 (1996).
Fulga, T. A., Elson-Schwab, I., Khurana, V., Steinhilb, M. L., Spires, T. L., Hyman, B. T. et al. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat. Cell Biol. 9, 139–148 (2007).
Heredia, L., Helguera, P., de Olmos, S., Kedikian, G., Sola Vigo, F., LaFerla, F. et al. Phosphorylation of actin-depolymerizing factor/cofilin by LIM-kinase mediates amyloid beta-induced degeneration: a potential mechanism of neuronal dystrophy in Alzheimer’s disease. J. Neurosci. 26, 6533–6542 (2006).
Beissbarth, T. & Speed, T. P. GOstat: find statistically overrepresented gene ontologies within a group of genes. Bioinformatics 20, 1464–1465 (2004).
Acknowledgements
This work was partially supported by the National Natural Science Foundation of China (grant nos. 30871394, 61073136, 91029717, 81172842, 60932008 and 61172098), the Specialized Research Fund for the Doctoral Program of Higher Education of China (grant no. 20112302110040), the Fundamental Research Funds for the Central Universities (grant no. HIT.ICRST.2010 022) and the Fund of Heilongjiang Health Department (grant nos.2011-204 and 2011-251).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Li, J., Wang, L., Xu, L. et al. DBGSA: a novel method of distance-based gene set analysis. J Hum Genet 57, 642–653 (2012). https://doi.org/10.1038/jhg.2012.86
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2012.86
