
Abstract
Neutral lipid storage disease with myopathy (NLSDM) referred to those neutral lipid storage disease (NLSD) patients with myopathy but without ichthyosis. Recently, NLSDM has been attributed to mutations in the PNPLA2 gene. Until now, 19 patients with PNPLA2 mutations have been reported. In the present study, we describe the clinical and genetic findings in three Chinese patients with NLSDM. Sequence analysis of PNPLA2 gene was performed. In our patients we identified four novel mutations in the PNPLA2 gene including two splicing mutations. The identification and study of mutations found in PNPLA2 is also particularly important to define the clinical spectrum and genotype–phenotype correlations of the PNPLA2 gene.
Similar content being viewed by others


Main
Neutral lipid storage disease (NLSD) refers to a group of diseases characterized with accumulation of triglyceride in several organs and tissues.1, 2, 3 Diagnosis of NLSD is based on the vacuoles of neutral lipids within leukocytes in peripheral blood which is called Jordans' anomaly.2, 3 The CDS (Chanarin–Dorfman syndrome) (MIM 275630), also known as NLSD with ichthyosis, is defined as an NLSD with ichthyosis, often associated with mild myopathy, cirrhosis, growth retardation, ataxia, hearing loss and mild mental retardation.4, 5 CDS is caused by the mutations of the CGI-58 gene. Neutral lipid storage disease with myopathy (NLSDM: MIM 610717) referred to those NLSD patients with myopathy but without ichthyosis. Recently, NLSDM has been attributed to mutations in the PNPLA2 gene.3, 6
PNPLA2 is the rate-limiting lipolytic enzyme and catalyzes the initial step in triglyceride hydrolysis.1, 7, 8 Most individuals with PNPLA2 mutations show atypical CDS-like symptoms, with myopathy and involvement of other organs but without ichthyosis.1, 3, 6, 9 Until now, 19 patients with PNPLA2 mutations have been reported. However, more cases are needed to define the clinical spectrum and genotype–phenotype correlations of the PNPLA2 gene. Here, we report the clinical and genetic findings in three Chinese patients with NLSDM.
We investigated three patients from unrelated families with NLSDM confirmed by muscle biopsy. The study was approved by the Ethics Committee of Shandong University's Qilu Hospital, and informed consents were obtained from all the participants. Patient 1 was a 44-year-old man with nonconsanguineous parents. He first noticed difficulty in walking and lifting heavy objects at age 41. As time progressed, he began to complain of muscle pain, fatigue and progressively limb weakness. Muscle atrophy was noticed in his bilateral upper and lower limbs, especially in the biceps brachii muscles and triceps muscles. Muscle weakness and atrophy were asymmetrical and proximally dominant. Serum CK was elevated to 651 IU l−1 (normally <140 IU l−1). Patient 2, a 48-year-old woman with consanguineous parents, complained of progressive muscle weakness in both upper and lower limbs over the past 3 years. Both proximal and distal muscles were asymmetrically involved, but distal muscle weakness and atrophy were more severe. Serum CK was elevated to 2419 IU l−1. Patient 3, a 39-year-old man with consanguineous parents. At 38 years, he noticed weakness of his right upper limb and the limb weakness gradually worsened. Over the next year, he developed gradually progressive weakness in his other limbs. Neurological examinations demonstrated severe asymmetric muscle weakness and wasting in the proximal part of the upper limbs. Serum creatine kinase was elevated to 1785 IU l−1. Neck muscle involvement and fasciculation were noticed in all the three patients (Figure 1a). For all patients, the family history was carefully checked, and no family history of muscle disorders was found. The clinical and laboratory characteristics of all patients are summarized in Table 1.

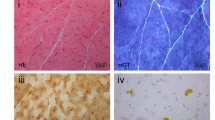
(a) The patients showed severe neck muscle and distal muscle atrophy. (b) Muscle biopsies of all patients show numerous lipid droplets in both types of fibers and Jordan's anomaly can be seen in the cytoplasm of a peripheral blood leukocyte of a patient. (c) Partial sequence of chromatogram of PNPLA2 gene in our patients. The arrows mark the mutations of PNPLA2. A full color version of this figure is available at the Journal of Human Genetics journal online.
Muscle biopsies were performed in all patients. Muscle biopsies revealed numerous lipid droplets in both types of fibers and most prominent in type one of the left biceps brachii muscles as well as rimmed vacuoles (Figure 1b). Peripheral blood leukocytes showed typical cytoplasmic lipid droplets (Jordan's anomaly). All the patients were diagnosed with NLSDM.
Genomic DNA was extracted from peripheral blood of all patients by standard methods. The coding exons and exon/intron boundaries of PNPLA2 gene were amplified using the primers described by Fischer et al.3 PNPLA2 has one transcript (Ref Seq ID: NM_020376.3). Direct sequencing of the coding exons and exon/intron boundaries of PNPLA2 gene revealed four distinct mutations in three patients (Figure 1c). Patient 1 was a compound heterozygote for a intronic mutation IVS6+2T>C, which changed the splice score from 0.37 to 0 in the donor splice site of intron 6, predicted using Neural Network software and a missense mutation c.749A>C that changes glutamine to proline at amino acid 250 (Q250P), which was not found in 50 healthy controls. Reverse transcription–PCR analysis detected only the c.749A>C mutant mRNA in skeletal muscle of the patient 1, indicating the absence of the other transcript resulted from the IVS6+2T>C splicing mutation. Patient 2 was homozygous for the mutation IVS6+1G>T, which changed the splice score from 0.37 to 0 in the donor splice site of intron 6. No PNPLA2 transcript was detected by RT–PCR of mRNA extracted from skeletal muscle of the patient 2. Patient 3 was homozygous for a single-base deletion (c.467del C). The deletion results in a frameshift beginning at amino acid 156. The frameshift also generates a premature stop codon at position 255. The mutation was thus designated as p.P156LfsX100.
Here, we report the clinical characteristics and genetic analysis of three patients with PNPLA2 gene mutations. Our patients showed initial symptoms of asymmetrical with PNPLA2 mutations. Similar findings were reported by Peter et al.1 And we performed an analysis of the clinical and genetic data of previously published cases of NLSDM (19 patients, see Supplementary Table 1). So asymmetrical may be common in primary symptoms of LSM with PNPLA2 mutations and it may be the first clue to NLSDM and help with the right diagnosis.
The N-terminal region of PNPLA2 contains a patatin-like phospholipase domain and GXSXG consensus sequence for serine lipases. The patatin domain (amino acids 10–179) contains an active site with a serine-aspartate catalytic dyad and an oxyanion hole that stabilizes the enzyme-substrate transition state.6, 10 The C-terminal region contains a potential lipid binding domain at residues 309–391.11 Most PNPLA2 mutations reported now affect the C-terminal hydrophobic domain, which is responsible for lipid binding. Only one patient carrying the PNPLA2 gene mutation c.475_478dupCTCC affected the patatin domain caused severely myopathy was reported.6 One of our patients carries the homozygous frameshift deletion mutation c.467delC, which may affect the patatin domain. Compared with other patients, the patient with the c.467delC mutation displayed similar phenotypes.1, 3, 6, 10, 11, 12 The result suggests that there is probably no correlation between the nature of the mutation in PNPLA2 and disease severity in NLSD. We also described two novel splicing mutations IVS6+1G>T and IVS6+2T>C of PNPLA2. This is the first report of splicing gene mutations of PNPLA2 gene. The missense mutation c.749A>C we found in patient 1 has not been described previously. It is unclear whether it is a pathogenic mutation. Patient 1 is definitely diagnosed with NLSDM. And all coding exons and exon/intron boundaries of PNPLA2 gene were sequenced. The mutation was also not detected in 50 controls. So we think this mutation is most likely a pathogenic mutation. The identification of more PNPLA2 mutations of NLSDM will lead to a more accurate diagnosis of the disease in patients as well as improved genetic counseling and prenatal diagnostic tests for couples who are carriers and planning a pregnancy. The identification and study of mutations found in PNPLA2 is also particularly important to help understand the mechanisms of the disease and develop more efficient and targeted therapies.
References
Reilich, P., Horvath, R., Krause, S., Schramm, N., Turnbull, D.M., Trenell, M. et al. The phenotypic spectrum of neutral lipid storage myopathy due to mutations in the PNPLA2 gene. J. Neurol. 258, 1987–1997 (2011).
Schweiger, M., Lass, A., Zimmermann, R., Eichmann, T.O. & Zechner, R. Neutral lipid storage disease: genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI-58/ABHD5. Am. J. Physiol. Endocrinol. Metab. 297, E289–E296 (2009).
Fischer, J., Lefevre, C., Morava, E., Mussini, J.M., Laforet, P., Negre-Salvayre, A. et al. The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nat. Genet. 39, 28–30 (2007).
Selimoglu, M.A., Esrefoglu, M., Gul, M., Gungor, S., Yildirim, C. & Seyhan, M. Chanarin-Dorfman syndrome: clinical features of a rare lipid metabolism disorder. Pediatr. Dermatol. 26, 40–43 (2009).
Bruno, C., Bertini, E., Di Rocco, M., Cassandrini, D., Ruffa, G., De Toni, T. et al. Clinical and genetic characterization of Chanarin-Dorfman syndrome. Biochem. Biophys. Res. Commun. 369, 1125–1128 (2008).
Akiyama, M., Sakai, K., Ogawa, M., McMillan, J. R., Sawamura, D. & Shimizu, H. Novel duplication mutation in the patatin domain of adipose triglyceride lipase (PNPLA2) in neutral lipid storage disease with severe myopathy. Muscle Nerve 36, 856–859 (2007).
Zimmermann, R., Lass, A., Haemmerle, G. & Zechner, R. Fate of fat: the role of adipose triglyceride lipase in lipolysis. Biochim. Biophys. Acta 1791, 494–500 (2009).
Zimmermann, R., Strauss, J.G., Haemmerle, G., Schoiswohl, G., Birner-Gruenberger, R., Riederer, M. et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 306, 1383–1386 (2004).
Akman, H.O., Davidzon, G., Tanji, K., Macdermott, E.J., Larsen, L., Davidson, M.M. et al. Neutral lipid storage disease with subclinical myopathy due to a retrotransposal insertion in the PNPLA2 gene. Neuromuscul. Disord. 20, 397–402 (2010).
Kobayashi, K., Inoguchi, T., Maeda, Y., Nakashima, N., Kuwano, A., Eto, E. et al. The lack of the C-terminal domain of adipose triglyceride lipase causes neutral lipid storage disease through impaired interactions with lipid droplets. J. Clin. Endocrinol. Metab. 93, 2877–2884 (2008).
Campagna, F., Nanni, L., Quagliarini, F., Pennisi, E., Michailidis, C., Pierelli, F. et al. Novel mutations in the adipose triglyceride lipase gene causing neutral lipid storage disease with myopathy. Biochem. Biophys. Res. Commun. 377, 843–846 (2008).
Ohkuma, A., Nonaka, I., Malicdan, M.C., Noguchi, S., Ohji, S., Nomura, K. et al. Distal lipid storage myopathy due to PNPLA2 mutation. Neuromuscul. Disord 18, 671–674 (2008).
Acknowledgements
We thank the patients and their families for their participation. This work was supported by the Research Award Fund for Outstanding Young Scientist of Shandong Province (Grant no. BS2012YY013) and the National Science Foundation Research Grant (Grant no. 81171182).
Web resources
Neural Network, http://www.fruitfly.org/seq_tools/splice.html
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Lin, P., Li, W., Wen, B. et al. Novel PNPLA2 gene mutations in Chinese Han patients causing neutral lipid storage disease with myopathy. J Hum Genet 57, 679–681 (2012). https://doi.org/10.1038/jhg.2012.84
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2012.84
Keywords
This article is cited by
-
Neutral lipid storage disease with myopathy in China: a large multicentric cohort study
Orphanet Journal of Rare Diseases (2019)
-
Novel PNPLA2 gene mutation in a child causing neutral lipid storage disease with myopathy
BMC Medical Genetics (2018)
