
Abstract
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative syndrome primarily affecting the upper and lower motor neurons. A characteristic neuropathological finding in ALS patients is neuronal inclusions positive for TAR DNA-binding protein 43 (TDP-43). Subsequently, mutations in the gene encoding TDP-43, TARDBP, proved to be involved in the development of ALS. We thus sequenced TARDBP in 177 Nordic ALS patients and found two previously reported (p.A90V and p.S379P) and two novel (p.G357R and p.R361T) missense variations in three familial ALS patients. The p.A90V and p.G357R variations were detected in the same patient and p.R361T was present in a family with both ALS and frontotemporal dementia-ALS. None of the missense variations were present in 200 neurologically healthy controls. However, p.A90V has also been reported in healthy individuals by others. Thus, the data suggest that these variations are rare and p.G357R, p.R361T and p.S379P are likely pathogenic but further functional characterization is needed to prove their pathogenicity. The mutation frequency in TARDBP in Nordic ALS patients was 1.7%. The ALS cohort was highly selected for a positive family history suggesting that mutations in TARDBP generally are a rare cause of ALS in Nordic countries.
Similar content being viewed by others

Introduction
Amyotrophic lateral sclerosis (ALS) is a heterogeneous group of neurodegenerative conditions which primarily affects the upper and/or lower motor neurons and their supporting cells.1, 2 However, ALS is often associated with cognitive impairment, which mainly includes executive functional behavior, verbal fluency and/or semantic functions indicating non-motor dysfunctions of the fronto-temporal lobes.1, 2, 3, 4 Depending on the neuropsychological tests used and the patient cohort, such cognitive impairments have been reported in a third to half the cases, and 5–15% of all ALS cases meet the diagnostic criteria for frontotemporal dementia (FTD).3, 5 The cognitive dysfunction may precede, follow or develop in parallel with the onset of motor symptoms. In all, 4–18% of all ALS patients have a self-reported family predisposition for the disease (FALS) with a mean age at onset between 48 and 52 years. This is about 10 years earlier than the mean age at onset in patients with SALS (sporadic ALS).1, 2
In 2006, reports on neuronal inclusions immunoreactive for truncated, hyperphosphorylated and ubiquitinated TAR DNA-binding protein 43 (TDP-43) in ALS and FTD patients gave reason to believe that mutations in the gene encoding TDP-43, TARDBP, could cause ALS and FTD.6, 7 Shortly after, studies found mutations segregating with disease, and functional studies supported a role for TDP-43 in motor neuron death.8, 9, 10, 11 To date, 45 TARDBP mutations have been reported in ALS patients (http://alsod.iop.kcl.ac.uk, http://bioinfo.hr/pro-mine/), the majority located in the glycine-rich region of exon 6. Furthermore, although rare, mutations in TARDBP have also been detected in patients with FTD-ALS and FTD.12, 13, 14 To determine the mutation frequency in TARDBP in the Nordic population, we sequenced 177 patients with ALS.
Materials and methods
Subjects
Genomic DNA from 177 patients diagnosed with ALS were selected from the ALS Biobank at Umeå University, Sweden. Preference was given to patients with a family history for ALS and/or dementia. In all, 30 patients were diagnosed as SALS and 147 patients were FALS with at least one more affected family member within three generations. Patients diagnosed as SALS did not fulfill the criteria for FALS or there was no information about the family history. Demographic data of the study cohort is summarized in Table 1. The ALS diagnosis was based on the El Escorial criteria.15 The diagnosis of ALS did not include systematic characterization of neuropsychological function. All patients had previously been screened negative for mutations in the genes superoxide dismutase 1 (SOD1), angiogenin (ANG), optineurin (OPTN), vesicle-associated membrane protein-associated protein b (VAPB) and progranulin (GRN). The control population consisted of 200 individuals with mini mental state examination scores⩾28 and without neurodegenerative diseases recruited from the Swedish National study on Aging and Care (http://www.snac-k.se/). When possible, DNA from blood relatives to the patients with a TARDBP variation were included for segregation analysis. This study was performed according to the tenants of the Declaration of Helsinki and with written informed consent for all participants and approval by local ethical review boards.
Mutation screening
TARDBP was screened for mutations by sequencing exons 1–5 and at least 20 flanking intronic nucleotides at the intron–exon boundaries. For exon 6, the coding region and approximately the first 780 bp of the 3′UTR were sequenced in all samples. Thereby, all known TDP-43 isoforms were screened for mutations. We also sequenced an additional approximately 840 bp downstream of position 780 bp in the 3′UTR in 120 patients. DNA was amplified using AmpliTaq Gold PCR Master Mix (Applied Biosystems, Branchburg, NJ, USA). The exons were sequenced in both directions with BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Austin, TX, USA) and analyzed on an ABI 3100 genetic analyzer. Primer sequences and PCR conditions are available upon request.
In silico analysis
The detected variations’ effect on splicing were analyzed by using the programs NetGene2 Server (http://www.cbs.dtu.dk/services/NetGene2/), Human Splicing Finder (http://www.umd.be/HSF/), GeneSplicer (http://cbcb.umd.edu/software/GeneSplicer/) and NNSPLICE (http://www.fruitfly.org/seq_tools/splice.html). The prediction programs PolyPhen (http://genetics.bwh.harvard.edu/pph/index.html) and SNAP (http://www.rostlab.org/services/SNAP/) were used to elucidate the missense variations’ effect on protein structure and function. For all prediction programs the default settings were used.
Results
Clinical summary and segregation analysis for the detected variations
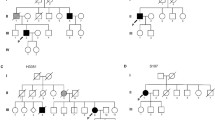
Nine genetic variations, four missense (p.A90V, p.G357R, p.R361T and p.S379P) and five non-coding (c.543+59A>G, c.*208G>A, c.*620A>G, c.*1008T>G and c.*1622A>T), and previously reported common single nucleotide polymorphisms (data not shown) were detected and are summarized in Table 2. A complete clinical description of the patients carrying the potentially pathogenic coding and non-coding variations and their family members can be found in the Supplementary text 1. A summary of the clinical information for the patients who carried the missense variations is presented in Table 3. Neuropathology was not available for any of patients with a detected variation.
The missense variations p.A90V and p.G357R were present in the same patient, 1:A, who developed bulbar onset ALS with dysarthria at age 67 years. A few months later, the patient developed muscle wasting and paresis in all four extremities. There were no signs of cognitive dysfunctions. The patient died of respiratory insufficiency 26 months after onset of symptoms. As patient 1:A was compound heterozygous for the variations p.A90V and p.G357R, we performed a haplotype analysis in six healthy family members, of whom four were above the age of 70 years. The data showed that the two variations were not present on the same chromosome. Two relatives between ages 50 and 70 years carried one of the variations and there was one healthy family member above 70 years who carried both missense variations. Unfortunately, no samples were available from any of the family members affected by ALS (the patient’s mother, uncle and a sibling) or the patient’s neurologically healthy father.
The missense variation p.R361T was identified in patient 2:A who developed spinal onset ALS with paresis in the left leg at the age of 69 years. At the end of the disease, the patient was tetraparalytic and had dysphagia and dysarthria. The patient died at the age of 75 years. DNA from six family members of the ALS patient heterozygous for p.R361T was available for segregation analysis. Of these, one sibling (patient 2:B) was diagnosed with FTD-ALS and five were healthy (between ages 40 and 75 years). p.R361T was present in patient 2:B but not in any of the healthy relatives. Patient 2:B, developed cognitive dysfunction, such as deficits in verbal fluency and memory, and was thus diagnosed with FTD at the age of 66 years. The patient also developed behavioral changes, expressive aphasia and became more temperamental and irritable. Furthermore, electromyography examination revealed signs of ALS and the patient had fasciculations but did not have paresis or muscle wasting. Before 2:B died at the age of 68 years, the patient had severe weight loss and generalized fasciculations in skeletal muscles and was diagnosed as FTD-ALS.
The p.S379P was present in patient 3:A, who developed ALS at the age of 62 years. The initial symptoms were weakness of the legs and walking difficulties. Later the muscle weakness and muscular atrophy also included the arms. The patient died 3 years after disease onset. There was a positive family history of ALS but no additional family members were available for segregation analysis.
Of the five non-coding variations detected in our study, similar segregation analyses were possible for two of the variations: c.543+59A>G and c.*208G>A. The c.543+59A>G was detected in patient 4:A and sequencing of seven additional family members, including a relative diagnosed with ALS, demonstrated that the variation was present in one of the healthy individuals but not in the other ALS patient. The non-coding c.*208G>A variation was detected in 16 patients of which one, a SALS patient, was homozygous for this variation. Segregation analysis in relatives to three patients who were heterozygous for c.*208G>A showed that this variation was present and absent in both affected patients and among healthy relatives.
Sequence analysis of exon 3, intron 4 and part of exon 6 in 200 cognitively healthy controls without neurodegenerative conditions, did not identify the missense variations or the non-coding variations c.543+59A>G and c.*620A>G (Table 2).
Bioinformatics
The coding variations p.G357R, p.R361T and p.A90V were predicted to affect the TDP-43 structure and function by at least one of the data prediction programs PolyPhen and SNAP. The p.S379P was predicted by both programs to be benign or neutral. These four variations showed high conservation when comparing the human amino acid sequence with six other species (Figure 1). None of the coding variations were predicted to result in splice site changes.

Multiple sequence alignment of TAR DNA-binding protein 43 in seven different species including Homo sapiens. The black area indicates the position of the identified missense variations and their corresponding position in other species. The gray areas indicate the amino acid substitutions in other species that do not correspond to the reference amino acid sequence in Homo sapiens. Protein accession number: aNP 031401.1; bXP 514380.2; cNP 663531.1; dNP 001026049.1; eNP 958884.1; fNP 477400.1; gXP 309663.4.
The non-coding variations’ effect on splicing was analyzed by using the programs NetGene2 Server, Human Splice Finder, GeneSplicer and NNSPLICE. The c.543+59A>G variation in intron 4 of TARDBP was suggested to create a new splice donor site in three of the prediction programs used (NetGene2 Server, Human Splice Finder and NNSPLICE) (data not shown). This new splice donor site was predicted to result in an 18 amino acid longer exon 4-coded protein and end with a premature stop codon, thus excluding exon 5 and 6. The other four non-coding variations (c.*208G>A, c.*620A>G, c.*1008T>G and c.*1622A>T) were not predicted to result in any specific structural or splice site changes (data not shown).
Discussion
Sequencing TARDBP in 177 patients with ALS resulted in the identification of four missense variations in three FALS patients and five non-coding variations. The missense variations were absent in 200 neurologically healthy controls. Patient 1:A was compound heterozygous for p.A90V and p.G357R, which were located on different chromosomes. The p.A90V has previously been reported in control individuals at a higher frequency compared with patients, suggesting that it is non-pathogenic.9, 10, 16, 17 However, p.A90V is located in the nucleus localization signal, which is conserved in several species. In the normal situation, the majority of TDP-43 is located in the nucleus. In an in vitro study, it was shown that p.A90V partially disrupted the normal distribution of TDP-43, that is, increased the cytoplasmic TDP-43 levels, indicating that this amino acid substitution may impair the shuttling of TDP-43 between the nucleus and the cytoplasm.17 The p.G357R has previously not been reported. The G357 is conserved between species and using the protein prediction programs PolyPhen and SNAP p.G357R was predicted to be pathogenic. So, it is possible that in this family the disease is caused by p.G357R, and that p.A90V contributes to the disease as a risk factor. Even though p.A90V is located in the nucleus localization signal, it is not enough to result in ALS by itself, which is demonstrated by the fact that there are reports on healthy control carries of this variation. Furthermore, there was a healthy relative to patient 1:A, who was above 70 years and also compound heterozygous for these two variations. This could be due to reduced penetrance of p.G357R, which is in agreement with other reported TARDBP mutations.8, 9, 10, 11, 12, 18
The third detected missense variation, p.R361T, was found in two siblings, one diagnosed with ALS and the other with FTD-ALS, and absent in five healthy family members supporting its pathogenic role in both ALS and FTD-ALS.
The fourth variation, p.S379P, detected in an FALS patient without dementia, is likely pathogenic, as it is located in a conserved residue and has been reported in a spinal onset FALS patient but not in 771 neurologically healthy controls.18
In the non-coding regions of TARDBP five genetic variations were detected, of which one (c.*208G>A) previously has been reported.12 Segregation analyses were performed for c.*208G>A and c.543+59A>G, and it suggested that neither is pathogenic. Similar to the previous reports, c.*208G>A was present in the control population. Of these five genetic variations in the non-coding region, only the c.543+59A>G in intron 4 was predicted to result in a new splice donor site by three of four splice site prediction programs. As no sample source for RNA or protein isolation was available, its effect on the RNA and protein levels could not be determined.
In total, three likely pathogenic mutations were detected in the Nordic ALS population, which corresponds to a mutation frequency of 1.7%. This is within the range of what has been estimated in other ALS populations, 1–3%.8, 9, 10, 11, 16, 17, 18, 19 However, the ALS patients in our study were intentionally selected for a positive family history suggesting that mutations in TARDBP are rare in the Nordic ALS population.
Our results give further support to shared genetic etiologies in some cases of FTD and ALS and continued studies are needed to be able to translate these findings into clinically useful diagnostic tools and treatment strategies.
Accession codes
References
Wijesekera, L. C., Leigh, P. N. Amyotrophic lateral sclerosis. Orphanet. J. Rare Dis. 4, 3 2009.
Andersen, P. M., Borasio, G. D., Dengler, R., Hardiman, O., Kollewe, K., Leigh, P. N. et al. Good practice in the management of amyotrophic lateral sclerosis: clinical guidelines. An evidence-based review with good practice points. EALSC Working Group. Amyotroph. Lateral Scler. 8, 195–213 2007.
Lomen-Hoerth, C., Anderson, T., Miller, B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 59, 1077–1079 2002.
Phukan, J., Pender, N. P., Hardiman, O. Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol. 6, 994–1003 2007.
Strong, M. J., Grace, G. M., Freedman, M., Lomen-Hoerth, C., Woolley, S., Goldstein, L. H. et al. Consensus criteria for the diagnosis of frontotemporal cognitive and behavioural syndromes in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 10, 131–146 2009.
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H. et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611 2006.
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T. et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 314, 130–133 2006.
Gitcho, M. A., Baloh, R. H., Chakraverty, S., Mayo, K., Norton, J. B., Levitch, D. et al. TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol. 63, 535–538 2008.
Kabashi, E., Valdmanis, P. N., Dion, P., Spiegelman, D., McConkey, B. J., Vande Velde, C. et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 40, 572–574 2008.
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B. et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 319, 1668–1672 2008.
Yokoseki, A., Shiga, A., Tan, C. F., Tagawa, A., Kaneko, H., Koyama, A. et al. TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann. Neurol. 63, 538–542 2008.
Benajiba, L., Le Ber, I., Camuzat, A., Lacoste, M., Thomas-Anterion, C., Couratier, P. et al. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann. Neurol. 65, 470–473 2009.
Borroni, B., Bonvicini, C., Alberici, A., Buratti, E., Agosti, C., Archetti, S. et al. Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum. Mutat. 30, E974–E983 2009.
Borroni, B., Archetti, S., Del Bo, R., Papetti, A., Buratti, E., Bonvicini, C. et al. TARDBP mutations in frontotemporal lobar degeneration: frequency, clinical features, and disease course. Rejuvenation Res. 13, 509–517 2010.
Brooks, B. R. El Escorial world federation of neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on motor neuron diseases/amyotrophic lateral sclerosis of the world federation of neurology research group on neuromuscular diseases and the El Escorial ‘Clinical limits of amyotrophic lateral sclerosis’ workshop contributors. J. Neurol. Sci. 124 (Suppl), 96–107 1994.
Guerreiro, R. J., Schymick, J. C., Crews, C., Singleton, A., Hardy, J., Traynor, B. J. TDP-43 is not a common cause of sporadic amyotrophic lateral sclerosis. PLoS One 3, e2450 2008.
Winton, M. J., Van Deerlin, V. M., Kwong, L. K., Yuan, W., Wood, E. M., Yu, C. E. et al. A90V TDP-43 variant results in the aberrant localization of TDP-43 in vitro. FEBS Lett. 582, 2252–2256 2008.
Corrado, L., Ratti, A., Gellera, C., Buratti, E., Castellotti, B., Carlomagno, Y. et al. High frequency of TARDBP gene mutations in Italian patients with amyotrophic lateral sclerosis. Hum. Mutat. 30, 688–694 2009.
Gijselinck, I., Sleegers, K., Engelborghs, S., Robberecht, W., Martin, J. J., Vandenberghe, R. et al. Neuronal inclusion protein TDP-43 has no primary genetic role in FTD and ALS. Neurobiol. Aging. 30, 1329–1331 2009.
Acknowledgements
We are indebted to the patients and their families for their participation in this project. Furthermore, Sabine Björk for her help with compiling clinical data and Ann-Charloth Nilsson for mutation analysis in other ALS genes. We also thank Laura Fratiglioni for providing the control samples. This project has been supported by Swedish Brain Power, Gun and Bertil Stohne's foundation, Gamla tjänarinnors foundation, Swedish Alzheimer foundation, Marianne & Marcus Wallenberg foundation, Knut and Alice Wallenberg foundation, Swedish Research Council, Swedish Brain Research Foundation, Hållstens Research Foundation and Swedish association for the neurologically disabled.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Supplementary information
Rights and permissions
About this article
Cite this article
Chiang, HH., Andersen, P., Tysnes, OB. et al. Novel TARDBP mutations in Nordic ALS patients. J Hum Genet 57, 316–319 (2012). https://doi.org/10.1038/jhg.2012.24
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2012.24
Keywords
This article is cited by
-
Nuclear-import receptors as gatekeepers of pathological phase transitions in ALS/FTD
Molecular Neurodegeneration (2024)
-
Genetics screening in an Italian cohort of patients with Amyotrophic Lateral Sclerosis: the importance of early testing and its implication
Journal of Neurology (2024)
-
A novel S379A TARDBP mutation associated to late-onset sporadic ALS
Neurological Sciences (2019)
-
The Role of MicroRNAs in Patients with Amyotrophic Lateral Sclerosis
Journal of Molecular Neuroscience (2018)
-
Genetic mutations in RNA-binding proteins and their roles in ALS
Human Genetics (2017)
