
Abstract
The mutation pattern of mitochondrial DNA (mtDNA) in mainland Chinese patients with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS) has been rarely reported, though previous data suggested that the mutation pattern of MELAS could be different among geographically localized populations. We presented the results of comprehensive mtDNA mutation analysis in 92 unrelated Chinese patients with MELAS (85 with classic MELAS and 7 with MELAS/Leigh syndrome (LS) overlap syndrome). The mtDNA A3243G mutation was the most common causal genotype in this patient group (79/92 and 85.9%). The second common gene mutation was G13513A (7/92 and 7.6%). Additionally, we identified T10191C (p.S45P) in ND3, A11470C (p. K237N) in ND4, T13046C (p.M237T) in ND5 and a large-scale deletion (13025–13033:14417–14425) involving partial ND5 and ND6 subunits of complex I in one patient each. Among them, A11470C, T13046C and the single deletion were novel mutations. In summary, patients with mutations affecting mitochondrially encoded complex I (MTND) reached 12.0% (11/92) in this group. It is noteworthy that all seven patients with MELAS/LS overlap syndrome were associated with MTND mutations. Our data emphasize the important role of MTND mutations in the pathogenicity of MELAS, especially MELAS/LS overlap syndrome.
Similar content being viewed by others


Introduction
Mitochondrial disorders, a group of diseases with defects in oxidative phosphorylation, are characterized by pleomorphic clinical manifestations, affecting multiple tissues and organs with a high energy demand. Important advances have been made regarding the genetic background of mitochondrial disorders in the past two decades.1, 2 Both primary alterations of the mitochondrial DNA (mtDNA) and mutations in nuclear genes affecting mitochondrial function have been identified as genetic causes of mitochondrial disease, as mitochondrial activity is a product of dual genetic control.3 The human mitochondrial genome is a 16 569 bp double-stranded circular DNA encoding for the 13 essential polypeptides of the oxidative phosphorylation system and the necessary RNA machinery (two ribosomal RNAs and 22 transfer RNAs (tRNAs)).4 There are three major types of pathological mtDNA mutations: large-scale rearrangements, point mutations in tRNAs or ribosomal RNAs and point mutations in protein-coding genes.
Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS), as one of the most commonly recognized mitochondrial disorders, has been reported worldwide.5, 6, 7, 8, 9 The clinical hallmark of MELAS is stroke-like episodes that cause the abrupt onset of focal neurological deficits at a young age. Neuroimaging studies and measurement of blood lactate are useful screening tests for the diagnosis of MELAS, though the definite diagnosis of MELAS relies on the ragged red fibers (RRFs) or strongly succinate dehydrogenase-stained vessels (SSVs) demonstrated by muscle biopsy, or the causative mtDNA mutation confirmed by molecular genetic studies.10 In 1990, Goto et al.5 first identified the mtDNA A3243G mutation as the molecular basis of MELAS, which was shown to account for more than 80% of the MELAS patients.11 However, at least 20 other distinct pathogenic mtDNA mutations have been reported in the other 20% patients with MELAS in the past two decades,12 suggesting great heterogeneity in the genotype of MELAS. Among them, the mtDNA 3271T>C point mutation was reported to be the second-most common mutation in Japanese MELAS patients.13, 14 Most reported MELAS mutations are located in the mitochondrial tRNA gene, but mutations in polypeptide-coding genes, including subunits 1, 5 and 6 of complex I (ND1, ND5 and ND6), have also been identified in MELAS or MELAS overlapping syndrome.12
As MELAS has such great genetic diversity, the investigation of mutation patterns will be very valuable in facilitating molecular diagnostic work-ups. Although the A3243G mutation is the most prevalent MELAS mutation, recent studies showed the mtDNA A3243G point mutation only appeared in 23% of Korean MELAS patients.9 Furthermore, the T3271C mutation was rarely reported in other countries than Japan.7, 15 These data suggest that the mutation pattern of MELAS can be different in different geographically localized populations. However, the mutation pattern of mtDNA is not yet clear in Chinese MELAS patients, with only several studies focusing on common mtDNA mutation screening.15, 16 Herein, we report the mtDNA mutation pattern, as well as three novel mtDNA mutations revealed by comprehensive mitochondrial genome mutation analysis, in a large cohort of unrelated Chinese patients with MELAS.
Materials and methods
Patients
From 1999 to 2010, 248 unrelated patients of Mainland Chinese descent were referred to the department of Neurology, Peking University First Hospital, to undertake muscular pathological exams or genetic tests for confirmation of a diagnosis of mitochondrial disorders. Among them, 92 patients with suspected MELAS who had complete clinical data were recruited for this study. All 92 suspected MELAS patients had at least one stroke-like episode without common vascular risk factors, which was confirmed by the corresponding lesions in the occipital, temporal, parietal and frontal lobes found by neuroimaging examination. Additional features include encephalopathy-like seizures or cognition decline, recurrent headaches or vomiting, elevated serum lactate, RRFs and/or SSVs on muscle pathology. mtDNA mutation screening and muscle biopsies were performed after informed consent was obtained. The study was undertaken with the approval of the institutional review board of our institute.
The clinical manifestations, laboratory data, neuroradiological changes and muscle pathological findings of 92 patients with MELAS are shown in Supplementary Table 1. Those patients with uncommon mtDNA mutations are described in detail below.
Patient 1
This 13-year-old girl was normal until the age of 6, when she developed painless vision loss in both eyes suddenly. A diagnosis of retrobulbar neuritis was made and her vision recovered to almost normal after 1 month. At age 10, she was found to have concomitant exotropia and abnormal gait. Brain magnetic resonance imaging (MRI) revealed symmetrical lesions in bilateral lenticular nucleus, thalamus and midbrain. At age 13, she suffered from seizures and right homonymous hemianopia following throbbing headache. Physical examination also showed short stature, hirsutism and ankle contractures. A new lesion in the left occipital lobe was disclosed by brain MRI. Blood lactate was 4.1 mmol l−1. Muscle biopsy revealed SSVs without RRFs or cytochrome c oxidase (COX)-negative fibers.
Patient 2
This female patient was a 19-year old who was well until the age of 11 years, at which time she developed recurrent migraines with auras. From age 16, she began to suffer from refractory myoclonus epilepsy in the right hand and generalized tonic/clonic seizure. At age 19, she suddenly developed blurred vision with slight hearing loss. On examination, she had cortical blindness, right-slanted tongue and right Babinski's sign. A cranial MRI showed high T2 signal lesions involving the bilateral parietal and occipital lobes, as well as symmetrical basal ganglia and midbrain lesions. Fundi fluorescence angiography was unremarkable. Muscle biopsy of the left biceps revealed no RRFs, COX-negative fibers or SSVs. Her family history was uninformative.
Patient 3
This female patient had slightly delayed psychomotor development and poor school performance from childhood. Since age 8, she was diagnosed as having nephropathy. At age 24, she was found to have left renal atrophy. Hearing loss was observed since age 25. From age 26 she developed focal seizures. At age 27, the patient had a stroke-like episode with high signals of T2 flair in the bilateral parietal and occipital lobes revealed by brain MRI. At age 28, she was hospitalized again because of another stroke-like episode. Physical examination revealed short stature, cortical blindness, nystagmus and right-sided hemianesthesia. Brain MRI disclosed multiple high T2-signals in bilateral parietal lobes, the left occipital lobe, as well as the basal ganglia and midbrain. Her condition deteriorated gradually and her parents discontinued therapy. Muscle biopsy showed RRFs and COX-positive SSVs. Her family history was negative.
Patient 4
This 30-year-old female patient had delayed growth development from childhood. At age 13, she developed painless vision loss in both eyes simultaneously over the course of a few days. There was little recovery after corticosteroid therapy. She remained otherwise well until age 20, when she developed ophthalmoplegia and ptosis. At age 21, she presented with episodes of generalized tonic-clonic seizures. A brain MRI revealed bilateral symmetrical high signals of the lenticular nucleus and midbrain on T2-weighted scans. From the age of 25, she developed progressive muscle weakness, frequent epileptic seizures, dysarthria and cognitive decline. A repetitive brain MRI at age 28 disclosed lesions with high T2 signals involving the bilateral temporal, parietal and occipital lobes, as well as periaqueductal regions in the midbrain. Muscle biopsy showed RRFs and SSVs. The family history was negative.
Patients 5, 6 and 7
The clinical details of these patients have been reported previously (patients 5, 6 and 7 as patient 1, 2 and 3, respectively).17 All of them presented with MELAS/Leigh syndrome (LS) overlap syndrome, based on both clinical manifestations and neuroimaging findings. None of them had a family history. Muscle biopsies from patients 5 and 7 showed RRFs and SSVs. Neither RRF, COX-deficient fiber nor SSV was found in muscle tissue from patient 6.
Patient 8
This male patient had a transient disturbance of consciousness after a car accident at the age of 21. After 1 month, he developed headaches in the left temporal region associated with hemianopia, agnosia and alexia. Brain CT showed low density in the left occipital lobe. After 5 months, he suffered from rightside headaches associated with numbness and weakness of the left arm. At age 22, he suffered third stroke-like episode. From age 25, he began to suffer from refractory focal and generalized tonic/clonic seizures. Brain MRI showed multiple lesions involving bilateral occipital, temporal and parietal lobes. Muscle biopsy at age 21 of the right biceps revealed several SSVs without RRFs or COX-negative fibers.
Patient 9
A 42-year-old man was well until the age of 19, when he developed painless loss of central vision in both eyes simultaneously over the course of a few days. A diagnosis of retrobulbar neuritis was made at that time. His vision recovered gradually, though not fully, in about 6 months. He developed sensorineural deafness in his twenties. At age 34, he received an operation for cataracts in both the eyes. He remained stable until age 39 when he had a stroke-like episode of motor aphasia, right hemiparesis and hemiparesthesia. A brain MRI showed high T2 signal in his left parietal and right frontal lobes. Muscle biopsy showed RRFs and COX-positive SSVs. He had further stroke-like episodes manifesting as left hemiparesis at age 40. From age 39, he had frequent seizures, which were refractory to anticonvulsants. Cognitive function declined progressively since the first stroke-like episode. He had a positive family history, with one of his younger sisters having several stroke-like episodes and cognitive decline, and his mother having hearing loss in her 50s.
Patient 10
A 36-year-old female reporter was well until age 30, when she was diagnosed as having hyperthyroidism because of palpitations and fatigue. She was given thiamazole and subsequently recovered. At age 33, she had an episode of right-side throbbing headache with nausea. After 1 week, she developed left hemiparesis following a generalized epileptic seizure. Cranial MRI showed high signal areas in the left parietal and right temporal lobes on T2-weighted scans. RRFs and SSVs were found by muscle biopsy. Until the time of this study, she had no further stroke-like episode. There was no family history of neurological disease.
Patient 11
This 32-year-old female patient had short stature and mild deafness from childhood. She was otherwise well until age 29, when over the course of a few days she developed right-side headache, blurred vision and numbness of the left extremities. Cranial MRI revealed lesions in the right parieto-occipital lobes. The symptoms resolved completely after 4 weeks of treatment. RRFs and SSVs were found by muscle biopsy performed at age 29. At age 30, she experienced another episode, with left-side headache, blurred vision and numbness of the right extremities. Although she improved after this second stroke-like episode, she developed focal motor seizures affecting her right arm and leg, which were responsive to anticonvulsants. There was no family history of neurological disease.
mtDNA analysis
Total DNA was extracted from blood, urinary sediment and muscle biopsy specimens using a standard procedure.18 For mutation analysis, first, the DNA samples were analyzed for the presence of large deletions and five common point mutations (A3243G, T3271C, A8344G, T8993G and T8993C) by southern blot/long-range PCR and PCR-restriction fragment-length polymorphism, respectively, according to published procedures.15, 19 Second, for those patients without common mutations, the entire mitochondrial genome extracted from muscle was sequenced by direct DNA sequencing of the PCR product using a BigDye terminator cycle sequencing kit (Applied Biosystem, Foster City, CA, USA) and an ABI 3730XL (Applied Biosystem) automated sequencer. We applied the long PCR-based sequencing method to avoid nuclear pseudogene amplification. The PCR products were then sequenced with 96 primer sets. The sequenced entire mtDNA was compared with a human mitochondrial genome database.20 Third, restriction fragment-length polymorphism analysis was used to determine the degree of heteroplasmy of the novel or uncommon point mutations. Fourth, we used Swiss-model PDBviewer software (http://swissmodel.expasy.org/) to predict possible changes in hydrophobicity and changes in configuration of transmembrane domains of the subunits due to the novel sequence variations. Finally, the novel mutations found in this study were screened in 300 normal subjects.
Results
Clinical features
Based on the clinical assessments as well as neuroimaging findings, this MELAS patients group (Supplementary Table 1) can be divided into two subgroups. Patients 1–7 were consistent with MELAS/LS overlap syndrome, presenting with ataxia, ophthalmoplegia, dysarthria, epilepsy and stroke-like episodes. Brain MRIs were characterized by the lesions distributed both in the cerebral cortexs as well as in the basal ganglia and brainstem symmetrically. Patients 8–92 showed symptoms in accordance with typical MELAS syndrome, manifesting as stroke-like episodes and additional features including hearing loss, diabetes mellitus, hirsutism, dementia and seizures.
Muscle pathology
Of 56 patients whose thorough myopathological data were available, both RRFs and SSVs could be seen in the muscle biopsies of 31 patients. In 13 patients, RRFs was detected but without SSV. In seven patients, only SSVs but no typical RRF was detected. In the remaining five patients, neither RRF nor SSV was found.
Mutation pattern
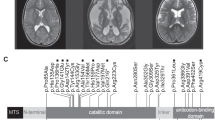
Genetic diagnosis was reached in all of the cases with MELAS (Supplementary Table 1). The A3243G mutation, as in other reports on MELAS, was the most common causal genotype in this patient group (79/92 and 85.9%). The T3271C mutation, which was the second-most common mutation in Japanese patients with MELAS,13, 14 was detected in only one patient (1/92 and 1.1%) in our study. A third tRNA Leu(UUR) mutation, T3291C, was detected in one other patient. In total, tRNA mutations accounted for 88.0% of the patients in this Chinese MELAS group.
In patient 3, long-range PCR of muscle mtDNA detected a 1.4 kb deletion. Further analysis showed that this deletion region spans from mtDNA np position 13025–13033/ to 14417–14425/, encompassing structural genes encoding partial ND5 and ND6.
In the remaining 10 patients, whole mtDNA sequencing revealed all were associated with point mutations in the mitochondrially encoded subunits of complex I (MTND) genes, including seven with a previously reported G13513A in the MTND5 each, one with a previously reported uncommon T10191C in MTND3, one with a novel A11470C in MTND4 and one with a novel T13046C in MTND5.
In patient 1, a new MTND5 mutation, T13046C, resulting in p.M237T in ND5, was detected in blood (22%), muscle (50%) and urinary sediment (52%). The healthy mother did not have this mutation in blood or urinary sediment, suggesting for a de novo mutation. This mutation was not detected in 300 normal subjects. p.M237T mutation showed strong conservation throughout different species.
In patient 8, a new MTND4 mutation, A11470C, resulting in p. K237N in ND4, was detected in muscle (55%), but was undetected in blood. The healthy brother did not have this mutation in blood, urinary sediment and hair root, which is also suggestive of a de novo mutation. This mutation was not detected in 300 normal subjects. Furthermore, the A11470C mutation changes a positive lysine to a neutralized asparagine at site 237, which is located in the sixth membrane-spanning α-helix. K237 is completely conserved in different species (Figure 1). Swiss-model PDBviewer software predicted that the residue K237 is near the end of the sixth helical structure and interacts with a highly conserved A233 residue via a potential hydrogen bond. If this lysine mutates to asparagine in the 237 residue, the N237 will lose the long side chain and the interaction between N237 and A233 will strengthen due to the establishment of more hydrogen bonds, causing the increase in the distance between N237 and Y241, which does great damage to the continuity of the helix (Figure 2).

Multiple alignments of mutation-containing ND4 region in different species. Protein sequences were aligned using the CLUSTALW program. The substituted amino acid (in shadow) for the mutation p. K237N (mtDNA A11470C) in patient 8, shows high conservation.

The residue K237 was near the end of the sixth helical structure of MTND4 (a). The mutation p.K237N will enhance the interaction between N237 and A233 due to the establishment of more hydrogen bonds, thus causing the increase in the distance between N237 and Y241 (b).
Discussion
We detected pathogenic mtDNA mutations in every MELAS patient in the study group, by common mtDNA mutation scanning as well as comprehensive analysis of the entire mitochondrial genome. This result highly suggested that all patients with MELAS have associated mtDNA mutations. For the distribution of mutation types, A3243G was the most common pathogenic mutation in our patient group, which is in line with most previous reports. The A3243G mutation was also the most frequently encountered mtDNA mutation in a large series of patients with suspected mitochondrial disease.16, 21 The G13513A mutation in the MTND5 was the second-most common mutation of MELAS (7.6%) following A3243G. If considering the mutations of ND3, ND4 and ND5 subunits together, MTND genes will be the second hotspot mutations, accounting for 12% of the mtDNA mutations causing MELAS in this test group.
It is noteworthy that all seven patients with MELAS/LS overlap syndrome (patients 1–7) were identified with MTND mutations, which contributed to the high frequency of MTND mutations in the present MELAS group. Among them, four patients were associated with G13513A, the other three patients with rare or novel mutations. Recently, an increasing number of cases with oxidative phosphorylation disease have been reported with MTND mutations, and among them, the G13513A mutation in MTND5 is the most frequently reported.22, 23 Three cases with MELAS/LS overlap syndrome carrying the G13513A mutation have been reported previously.24, 25 Four cases with childhood/juvenile onset MELAS/LS overlap syndrome (patients 4–7) reported in this study again stress that the G13513A mutation should be screened preferentially in patients with MELAS/LS overlap syndrome. In addition to G13513A, at least 10 other MTND5 mutations have been reported to be associated with the oxidative phosphorylation disease.20, 26 In this study, we reported a new MTND5 point mutation, T13046C (p.M237T), which was also detected in a patient with typical MELAS/LS overlap syndrome (patient 1). Although functional studies of the new sequence variant have to be performed, we consider the T13046C to be the causative mutation in the patient 1 based on its heteroplasmy, strong conservation and absence in several tissues of the unaffected mother of the index patient. Furthermore, A13045C resulting in M237L was recently reported in a patient with MELAS/LS,27 who was very similar to our patient 1, which can serve as another evidence supporting the pathogenicity of T13046C.
Apart from the MTND5 gene, other MTND genes could be associated with MELAS/LS overlap syndrome as well.28, 29, 30, 31 In this study, we found T10191C in MTND3, and a large deletion (from 13025–13033/to 14417–14425/) encompassing partial MTND5 and MTND6 in patients 2 and 3, respectively. The T10191C mutation, detected in patient 2 in this study, was first reported in a patient that presented with a progressive Leigh-like clinical picture of epilepsy, strokes, optic atrophy and cognitive decline by Taylor et al.32 Subsequently, several MELAS or LS patients were reported carrying the T10191C mutation.31, 33, 34, 35 The clinical features of patient 2 in the present study conformed with typical MELAS but her brain MRI showed lesions in both cerebral cortex and midbrain, suggesting that a diagnosis of MELAS/LS overlap syndrome was more appropriate. Patient 3 presented a similar clinical picture as patient 2, with a final diagnosis of MELAS/LS overlap syndrome based on the combination of clinical manifestations and brain MRI findings. Further genetic analysis revealed a large-scale mtDNA deletion and no point mutations. So far, only one patient with MELAS has been reported to carry a large-scale deletion of mtDNA,36 which has been shown to be frequently responsible for CPEO, KSS and Pearson syndrome. The deletion we report in this study, also encompassing structural genes encoding partial ND5 and ND6 of complex I, is a novel deletion. These two cases further emphasize the role of mtDNA complex I mutations in the molecular pathogenesis of MELAS/LS overlap syndrome. Also, the MELAS patient with a novel single large-scale deletion adds to the complexity of the gene variation database of MELAS/LS overlap syndrome.
For the classic MELAS subgroup (patient 8–92), G13513A mutation of complex I still ranked the second-most common (3/85 and 3.5%), following A3243G mutation(79/85 and 93.0%). All these three patients manifested late-onset stroke-like episodes, similar to those G13513A causing MELAS patients reported by others.23, 24, 37, 38 In addition, a novel missense mutation, A11470C of complex I, was identified in an adult-onset MELAS patient (patient 10), in agreement with the notion that mutations of mitochondrial complex I genes have an important role in late-onset MELAS. Several aspects of the evidence can support etiologic role of the A11470C mutation. First, the diagnosis of MELAS can be firmly established in patient 10, according to the clinical picture and pathological findings. Second, sequencing of the entire mtDNA revealed no other pathogenic mutations, except for the A11470C substitution. The heteroplasmic mutation was confirmed in the patient by PCR-restriction fragment-length polymorphism, and the ratio of mutations was higher in postmitotic tissue (skeletal muscle) than in mitotic cells (circulating lymphocytes). This phenomenon is well recognized in many other pathogenic mtDNA point mutations. Third, the mutation was not only undetected in 300 normal subjects, but also has not been reported previously as a polymorphic variant in MITOMAP. This indicates that the A11470C mutation is pathogenic in the family. Fourth, although it is open to discussion whether ND4 has a direct role in electron transfer or proton translocation of the complex I subunit, another p.T109A mutation of ND4 has been identified in a typical MELAS patient.28 The p.K237 residue is highly conserved through different species. Furthermore, as predicted by the Swiss-model PDBviewer software, p. K237N would greatly influence the spatial structure of the ND4 subunit of complex I. Therefore, although functional studies to support the pathogenicity of the new sequence variant need to be performed, we consider the mutation A11470C in the ND4 gene to be the causative mutation in patient 10.
In this study, in every patient, we reached a molecular diagnosis, compared with the relatively lower detection rate in large-scale screening. We believe that the high level of molecular diagnosis is due to the following reasons: first, the establishment of the phenotype is very important. Other conditions may mimic MELAS,39 making the differential diagnosis difficult. A proper diagnosis should be based on a multidisciplinary approach that includes clinical work-up, biochemical analysis, radiological changes and pathological findings. Among them, RRFs and SSVs on pathological examination have a very important role in directing the diagnosis. Sometimes COX-positive SSV itself can be a very strong indicator of MELAS, even in the case of the absence of RRFs.40 In seven patients of the present study, only SSVs were found, but their diagnosis was confirmed by molecular analysis. Secondly, while muscle biopsy is very important, it cannot be used alone to diagnose MELAS.41 In our patient group, it is noteworthy that five patients showed neither RRF nor SSV on muscle pathology, making the diagnosis difficult initially. However, whole mtDNA mutational analysis identified that they harbored mtDNA mutations. Therefore, from a clinical and diagnostic standpoint, this work demonstrates that sequence analysis of the entire mtDNA is necessary in all cases with a suspect of mitochondrial etiology, even if not all clinical and diagnostic criteria are fulfilled. This recommendation is further corroborated by the recent evidence that mutations of mtDNA are far more frequent in the general population than previously suspected.42 Finally, attention should be paid to the tissue specimen chosen when doing mtDNA analysis. In this study, there were five patients whose molecular diagnosis were negative initially, when their blood was used for the genetic test. But mtDNA mutations were detected in their muscle samples or urinary sediments later (patient 3, 8–10 and 19). So tests on a blood sample may be negative, leading to a false negative diagnosis, especially in the case of a mutation with a low mutation load. Meanwhile, urinary sediment or follicles can be good non-invasive samples for mtDNA mutation screening.8, 18 Also, in order to avoid false negative results when doing whole mtDNA sequencing, muscle or other post-mitotic tissue DNA is highly recommended, especially when we take both the high cost and time consumption of whole mtDNA sequencing into consideration.
In conclusion, mutations affecting mitochondrially encoded complex I (MTND) reached unexpectedly high as 12.0% in this MELAS group, indicating the important role of MTND mutations in the pathogenicity of MELAS, especially MELAS/LS overlap syndrome. Our results also highlight that thorough clinical work-up and evaluation can provide a good platform for a highly specific molecular genetic analysis. Therefore, although costly and time-consuming, sequencing of the entire mtDNA is still necessary for patients with highly suspected MELAS but without common mutations, even in the absence of a specific histochemical abnormality.
References
McFarland, R., Taylor, R. W. & Turnbull, D. M. The neurology of mitochondrial DNA disease. Lancet Neurol. 1, 343–351 (2002).
DiMauro, S. & Schon, E. A. Mitochondrial respiratory-chain diseases. N. Engl. J. Med. 348, 2656–2668 (2003).
Zeviani, M. & Carelli, V. Mitochondrial disorders. Curr. Opin. Neurol. 16, 585–594 (2003).
Anderson, S., Bankier, A. T., Barrell, B. G., de Bruijn, M. H., Coulson, A. R., Drouin, J. et al. Sequence and organization of the human mitochondrial genome. Nature 290, 457–465 (1981).
Goto, Y., Nonaka, I. & Horai, S. Mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 348, 651–653 (1990).
Hirano, M. & Pavlakis, S. G. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS): current concepts. J. Child Neurol. 9, 4–13 (1994).
Jaksch, M., Kleinle, S., Scharfe, C., Klopstock, T., Pongratz, D., Müller-Höcker, J. et al. Frequency of mitochondrial transfer RNA mutations and deletions in 225 patients presenting with respiratory chain deficiencies. J. Med. Genet. 38, 665–673 (2001).
McDonnell, M. T., Schaefer, A. M., Blakely, E. L., McFarland, R., Chinnery, P. F., Turnbull, D. M. et al. Noninvasive diagnosis of the 3243A > G mitochondrial DNA mutation using urinary epithelial cells. Eur. J. Hum. Genet. 12, 778–781 (2004).
Choi, B. O., Hwang, J. H., Cho, E. M., Jeong, E. H., Hyun, Y. S., Jeon, H. J. et al. Mutational analysis of whole mitochondrial DNA in patients with MELAS and MERRF diseases. Exp. Mol. Med. 42, 446–455 (2010).
Sproule, D. M. & Kaufmann, P. Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes: basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome. Ann. N.Y. Acad. Sci. 1142, 133–158 (2008).
Goto, Y., Horai, S., Matsuoka, T., Koga, Y., Nihei, K., Kobayashi, M. et al. Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS): a correlative study of the clinical features and mitochondrial DNA mutation. Neurology 42, 545–550 (1992).
Ruiz-Pesini, E., Lott, M. T., Procaccio, V., Poole, J., Brandon, M. C., Mishmar, D. et al. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids. Res. 35, D823–D828 (2007).
Sakuta, R., Goto, Y., Horai, S. & Nonaka, I. Mitochondrial DNA mutations at nucleotide positions 3243 and 3271 in mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes: a comparative study. J. Neurol. Sci. 115, 158–160 (1993).
Chou, H. F., Liang, W. C., Zhang, Q., Goto, Y. & Jong, Y. J. Clinical and genetic features in a MELAS child with a 3271T>C mutation. Pediatr. Neurol. 38, 143–146 (2008).
Qi, Y., Zhang, Y., Wang, Z., Yang, Y., Yuan, Y., Niu, S. et al. Screening of common mitochondrial mutations in Chinese patients with mitochondrial encephalomyopathies. Mitochondrion 7, 147–150 (2007).
Cao, Y., Ma, Y., Zhang, Y., Li, Y., Fang, F., Wang, S. et al. Detection of eight frequently encountered point mutations in mitochondria in Chinese patients suggestive of mitochondrial encephalomyopathies. Mitochondrion 10, 330–334 (2010).
Wang, Z., Qi, X. K., Yao, S., Chen, B., Luan, X., Zhang, W. et al. Phenotypic patterns of MELAS/LS overlap syndrome associated with m.13513G>A mutation, and neuropathological findings in one autopsy case. Neuropathology 30, 606–614 (2010).
Ma, Y., Fang, F., Yang, Y., Zou, L., Zhang, Y., Wang, S. et al. The study of mitochondrial A3243G mutation in different samples. Mitochondrion 9, 139–143 (2009).
Akanuma, J., Muraki, K., Komaki, H., Nonaka, I. & Goto, Y. Two pathogenic point mutations exist in the authentic mitochondrial genome, not in the nuclear pseudogene. J. Hum. Genet. 45, 337–341 (2000).
MITOMAP. A Human Mitochondrial Genome Database, http://www.mitomap.org (2011).
Schaefer, A. M., McFarland, R., Blakely, E. L., He, L., Whittaker, R. G., Taylor, R. W. et al. Prevalence of mitochondrial DNA disease in adults. Ann. Neurol. 63, 35–39 (2008).
Hanna, M. G., Nelson, I. P., Morgan-Hughes, J. A. & Wood, N. W. MELAS: a new disease associated mitochondrial DNA mutation and evidence for further genetic heterogeneity. J. Neurol. Neurosurg. Psychiatry 65, 512–517 (1998).
Shanske, S., Coku, J., Lu, J., Ganesh, J., Krishna, S., Tanji, K. et al. The G13513A mutation in the ND5 gene of mitochondrial DNA as a common cause of MELAS or Leigh syndrome: evidence from 12 cases. Arch. Neurol. 65, 368–372 (2008).
Pulkes, T., Eunson, L., Patterson, V., Siddiqui, A., Wood, N. W., Nelson, I. P. et al. The mitochondrial DNA G13513A transition in ND5 is associated with a LHON/MELAS overlap syndrome and may be a frequent cause of MELAS. Ann. Neurol. 46, 916–919 (1999).
Pénisson-Besnier, I., Reynier, P., Asfar, P., Douay, O., Sortais, A., Dubas, F. et al. Recurrent brain hematomas in MELAS associated with an ND5 gene mitochondrial mutation. Neurology 55, 317–318 (2000).
Blok, M. J., Spruijt, L., de Coo, I. F., Schoonderwoerd, K., Hendrickx, A. & Smeets, H. J. Mutations in the ND5 subunit of complex I of the mitochondrial DNA are a frequent cause of oxidative phosphorylation disease. J. Med. Genet. 44, e74 (2007).
Liolitsa, D., Rahman, S., Benton, S., Carr, L. J. & Hanna, M. G. Is the mitochondrial complex I ND5 gene a hot-spot for MELAS causing mutations? Ann. Neurol. 53, 128–132 (2003).
Lertrit, P., Noer, A. S., Jean-Francois, M. J., Kapsa, R., Dennett, X., Thyagarajan, D. et al. A new disease-related mutation for mitochondrial encephalopathy lactic acidosis and strokelike episodes (MELAS) syndrome affects the ND4 subunit of the respiratory complex I. Am. J. Hum. Genet. 51, 457–468 (1992).
Ravn, K., Wibrand, F., Hansen, F. J., Horn, N., Rosenberg, T. & Schwartz, M. An mtDNA mutation, 14453G-A, in the NADH dehydrogenase subunit 6 associated with severe MELAS syndrome. Eur. J. Hum. Genet. 9, 805–809 (2001).
Kirby, D. M., McFarland, R., Ohtake, A., Dunning, C., Ryan, M. T., Wilson, C. et al. Mutations of the mitochondrial ND1 gene as a cause of MELAS. J. Med. Genet. 41, 784–789 (2004).
McFarland, R., Kirby, D. M., Fowler, K. J., Ohtake, A., Ryan, M. T., Amor, D. J. et al. De novo mutations in the mitochondrial ND3 gene as a cause of infantile mitochondrial encephalopathy and complex I deficiency. Ann. Neurol. 55, 58–64 (2004).
Taylor, R. W., Singh-Kler, R., Hayes, C. M., Smith, P. E. & Turnbull, D. M. Progressive mitochondrial disease resulting from a novel missense mutation in the mitochondrial DNA ND3 gene. Ann. Neurol. 50, 104–107 (2001).
Lebon, S., Chol, M., Benit, P., Mugnier, C., Chretien, D., Giurgea, I. et al. Recurrent de novo mitochondrial DNA mutations in respiratory chain deficiency. J. Med. Genet. 40, 896–899 (2003).
Malfatti, E., Bugiani, M., Invernizzi, F., de Souza, C. F., Farina, L., Carrara, F. et al. Novel mutations of ND genes in complex I deficiency associated with mitochondrial encephalopathy. Brain 130, 1894–1904 (2007).
Valente, L., Piga, D., Lamantea, E., Carrara, F., Uziel, G., Cudia, P. et al. Identification of novel mutations in five patients with mitochondrial encephalomyopathy. Biochim. Biophys. Acta. 1787, 491–501 (2009).
Campos, Y., Garcia-Silva, T., Barrionuevo, C. R., Cabello, A., Muley, R. & Arenas, J. Mitochondrial DNA deletion in a patient with mitochondrial myopathy, lactic acidosis, and stroke-like episodes (MELAS) and Fanconi's syndrome. Pediatr. Neurol. 13, 69–72 (1995).
Santorelli, F. M., Tanji, K., Kulikova, R., Shanske, S., Vilarinho, L., Hays, A. P. et al. Identification of a novel mutation in the mtDNA ND5 gene associated with MELAS. Biochem. Biophys. Res. Commun. 238, 326–328 (1997).
Corona, P., Antozzi, C., Carrara, F., D′Incerti, L., Lamantea, E., Tiranti, V. et al. A novel mtDNA mutation in the ND5 subunit of complex I in two MELAS patients. Ann. Neurol. 49, 106–110 (2001).
Ringelstein, E. B., Kleffner, I., Dittrich, R., Kuhlenbäumer, G. & Ritter, M. A. Hereditary and non-hereditary microangiopathies in the young. An update. J. Neurol. Sci. 299, 81–85 (2010).
Hasegawa, H., Matsuoka, T., Goto, Y. & Nonaka, I. Strongly succinate dehydrogenase-reactive blood vessels in muscles from patients with mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. Ann. Neurol. 29, 601–605 (1991).
Ujike, H., Wakagi, T., Kohira, I., Kuroda, S., Otsuki, S. & Sato, T. MELAS without ragged red fibers or lactic acidosis diagnosed by mitochondrial DNA testing. Jpn. J. Psychiatry Neurol. 47, 637–641 (1993).
Chinnery, P. F., Johnson, M. A., Wardell, T. M., Singh-Kler, R., Hayes, C., Brown, D. T. et al. The epidemiology of pathogenic mitochondrial DNA mutations. Ann. Neurol. 48, 188–193 (2000).
Acknowledgements
We thank all the doctors that referred patients to our department, as well as all patients and their family members that participated in this study. The authors also wish to thank Dr Sue Zhong for editorial assistance. This study was supported by the National Nature Science Foundation of China (No.30870864).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Zhao, D., Hong, D., Zhang, W. et al. Mutations in mitochondrially encoded complex I enzyme as the second common cause in a cohort of Chinese patients with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes. J Hum Genet 56, 759–764 (2011). https://doi.org/10.1038/jhg.2011.96
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2011.96
Keywords
This article is cited by
-
Clinical and pathological features in adult-onset NIID patients with cortical enhancement
Journal of Neurology (2020)
-
Mitochondrial dysfunction and endoplasmic reticulum stress involved in oocyte aging: an analysis using single-cell RNA-sequencing of mouse oocytes
Journal of Ovarian Research (2019)
