
Abstract
Emery–Dreifuss muscular dystrophy (EDMD) is a neuromuscular disorder exhibiting a cardiomyopathy with cardiac conduction defects. X-linked EDMD arises from mutations in the EMD gene, which encodes for a nuclear membrane protein termed emerin. In this study, we describe novel and recurrent EMD mutations identified in 18 probands and three carriers from a cohort of 255 North American patients referred for EDMD genetic mutation analysis. Eight of these mutations are novel including six frameshift mutations (p.D9GfsX24, p.F39SfsX17, p.R45KfsX16, p.F190YfsX19, p.R203PfsX34 and p.R204PfsX7) and two non-sense mutations (p.S143X, p.W200X). Our data augment the number of EMD mutations by 13.8%, equating to an increase of 5.2% in the total known EMD mutations and to an increase of 6.0% in the number of different mutations. Analysis of the exon distribution of mutations within the EMD gene, suggests a nonrandom distribution, with exon 2 as a hot spot. This phenomenon may be due to its high GC content, which at 60% is the most GC-rich exon in the EMD gene.
Similar content being viewed by others


Introduction
Emery–Dreifuss muscular dystrophy (EDMD) manifests in childhood with slowly progressive muscle weakness in a scapulo-humeroperoneal distribution, associated with contractures of the Achilles tendon, neck and elbow.1, 2 Patients typically remain ambulant for life. Cardiac involvement is a consistent feature, initially involving arrhythmias, progressing towards complete heart block with a substantial risk of sudden death in middle age. Both inter- and intra-familial variability in clinical symptomatology and age of onset occurs.3 Distinguishing EDMD from other overlapping phenotypes, such as Becker muscular dystrophy, limb-girdle muscular dystrophy, facioscapulohumeral muscular dystrophy (FSHD), spinal muscular dystrophy and rigid spine syndrome,4 can be difficult without genetic testing.5 Five genes have been associated with the EDMD phenotype (EMD, LMNA, FHL1, SYNE1 and SYNE2), but collectively they only account for 46% of EDMD patients,6 suggesting the existence of other genes involved in the development of this disorder.
Both X-linked and autosomal forms of EDMD occur, with the X-linked form being described first in 1966 by Emery and Dreifuss.7 This form of the disease is the milder of the two; with the first symptoms observed at a mean age of 5.6 years, and cardiomyopathy detected at a mean age of 30.5 years.8 Cardiac conduction defects are correctable by pacemaker insertion. Female carriers can develop the cardiomyopathy asymptomatically where they are at risk from sudden cardiac death. Two genes are associated with X-linked EDMD (X-EDMD, MIM #310 300), EMD9 and FHL1.10 Mutations in EMD are very rare, with an estimated incidence of 0.13/100 000.11 The human EMD gene is 2100 base pairs in length, containing six exons, with an open reading frame of 762 base pairs encoding a single-membrane spanning 254-residue protein termed emerin (Figures 1a and b; Bione et al.9). Structurally it has an N-terminal nucleoplasmic domain of 221 residues, followed by a transmembrane region and a C-terminal 11-residue tail. Emerin has an ubiquitous tissue distribution where it is anchored at the inner nuclear membrane.12 Two allelic disorders are known, X-linked limb-girdle muscular dystrophy13 and X-linked sinus node dysfunction.14, 15 The latter is notable for the lack of skeletal muscle involvement. Collectively, these diseases are known as the emerinopathies. Mutations in the four and a half LIM domain gene, FHL1, are associated with X-EDMD and several allelic disorders including reducing body myopathy, X-linked scapuloperoneal myopathy, X-linked myopathy with postural muscle atrophy and X-linked hypertrophic cardiomyopathy. In this instance, mutational–positional effects determine which disorder develops.10, 16

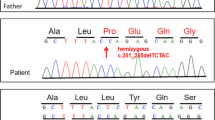
Schematic drawing of (a) human EMD cDNA and (b) human emerin protein structure and position of the mutations described in this study. (a) The EMD gene (GenBank X82434.1) contains 6 exons shown in dark shaded boxes joined by introns. Numbers above the exons refer to amino-acid numbers. The two splicing mutations are illustrated. (b) Emerin is shown with the LEM domain (residues 2–44), transmembrane (TM) domain (residues 221–244) and the poly-serine-rich region (residues 185–199). Mutations reported in this study are shown above, with the novel mutations in grey and the recurring in black text. The region encoded by exon 2 is enlarged to illustrate the functional motifs it contains. These include residues 28–44 of the LEM domain, the nuclear localisation sequence (NLS; residues 35–47) and two characterised phosphorylation sites, S49 (protein kinase A) and Y59 (Src tyrosine kinase). Drawn to scale. A full color version of this figure is available at the Journal of Human Genetics journal online.
The incidence of autosomal EDMD (A-EDMD) is unknown but is significantly more common than X-EDMD. The autosomal form of the disease is generally more severe than X-EDMD, with an average age of onset of 32 months and the cardiomyopathy developing in the early teens, but overall exhibiting a wider range of clinical severity.3, 17 Only one gene has been linked to typical A-EDMD (MIM #181 350): the LMNA gene (1q21.3; Bonne et al.18). Alternative splicing of LMNA produces the intermediate filament proteins, nuclear lamin A and C, which form the nuclear lamina on the inside of the inner nuclear membrane. Both autosomal dominant (EDMD2) and autosomal recessive (EDMD3) forms of A-EDMD occur. Non-surprisingly, the LMNA- and EMD-causing EDMD phenotypes overlap, given emerin and lamin A/C are binding partners.19 Interestingly, a congenital form of A-EDMD has recently been attributed to specific LMNA mutations (L-CMD).17 Atypical A-EDMD phenotypes, again with no skeletal muscle involvement, have been ascribed to the SYNE1 and SYNE2 genes that encode nesprin-1 and -2, respectively, which are emerin–lamin A/C-binding proteins.20
This study describes 21 novel and recurrent EMD gene mutations identified in a North American cohort, referred for genetic testing following a suspected diagnosis of EDMD. This is the first report of the distribution of EMD mutations from a large patient cohort from outside Europe. We report eight novel mutations including six frameshift mutations (p.D9GfsX24, p.F39SfsX17, p.R45KfsX16, p.F190YfsX19, p.R203PfsX34 and p.R204PfsX7) and two non-sense mutations (p.S143X and p.W200X). Our data extends the number of EMD mutations by 13.8%, equating to an increase of 5.2% in the total known EMD mutations and to an increase of 6.0% in the number of different mutations. Analysis of the distribution of mutations in the exons, taking into account exon size, identifies exon 2 as a hot spot. Complementary DNA sequence analysis suggests that this feature may be due to this exon's high-GC content.
Materials and methods
Patients
The patients (n=255) reported in this study were referred to either the DNA Diagnostic Laboratory at Carolinas Medical Center (1996–2001) or the Molecular Diagnostics Laboratory at the University of Minnesota (2001–2003) from the United States of America and Canada for mutation analysis in the EMD and LMNA genes. Patients were selected for genetic screening following a clinical diagnosis of EDMD, limb-girdle muscular dystrophy, Becker muscular dystrophy or FSHD. FHL1, SYNE1 and SYNE2 mutations were not screened, as the screening programme predated linkage of these genes to the EDMD phenotype. This study was approved by the Institutional Review Board at both Carolinas Medical Center and the University of Minnesota and each of the participating institutions sending blood or DNA samples under a research protocol.
DNA isolation
DNA was extracted from peripheral blood by either standard proteinase K/phenol-chloroform extraction procedures on a 341 Nucleic Acid Extractor (Perkin-Elmer, Norwalk, CT, USA) or using a QIAamp DNA Blood Mini Kit (Qiagen, Valencia, CA, USA).
PCR
The emerin gene was amplified in two fragments using primer sets 106F/484R for the amplification of exons 1 and 2, and primers 494F/1868R for the amplification of exons 3–6. Primer sequences are listed in Supplementary Table 1. The amplifications were performed in a 50 μl volume, containing 200 ng genomic DNA template; 1.5 mM MgCl2, 10 mM TRIS-HCL, pH 8.8, 50 mM KCl, 1 mM each dNTP, 10%DMSO, 1 μM each primer and 2.5 U Taq polymerase (Perkin-Elmer). PCR cycling conditions in a MJ Research PTC200 thermal cycler were: 95 °C for 10 min, followed by 35 cycles of 95 °C for 1 min, 60 °C for 1 min, 72 °C for 1 min and a final extension of 72 °C for 7 min.
Sequencing
Amplimers were extracted from agarose gel using a Gel Extraction kit (Qiagen) and each exon was sequenced directly from both strands using the Prism Ready Reaction DyeDeoxy Terminator Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) with analysis of sequenced products on either an ABI377 or ABI3100 Genetic Analyzer. Generated sequences were compared with the published EMD gene sequence using the Sequence Navigator (Applied Biosystems) or Sequencher (Gene Codes, Ann Arbor, MI, USA) software packages. Nucleotide numbering reflects complementary DNA numbering with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence, according to journal guidelines (http://www.hgvs.org/mutnomen). The initiation codon is codon 1.
Results
Clinical and genetic information on 23 referrals positive for EMD mutations
This is the first report on the distribution of EMD mutations from a large patient cohort from North America. Among the 255 referrals, 61 (23.9%) were found to have LMNA mutations21, 22 and a further 23 (9.0%) had EMD mutations. Clinical and genetic information on two of the patients with EMD mutations have been published previously (p.P183H;23 p.Q247LfsX110.24) and will not be described further here. One EMD patient (6784) additionally presented with a D4Z4 contraction at 4q35 (unpublished data, Meriggioli), suggesting that he additionally had FSHD.25 Of the remaining 21 referrals, three were identified as carrier females (722, 2370 and 1053) and 18 as patients (Figure 1b). In this patient cohort there were eight novel and 10 recurrent EMD mutations. One novel mutation (p.W200X) occurred in an unrelated proband (1196) and carrier female (722). Two recurrent mutations were each identified in two separate probands (p.M1? in 6784 and 921; p.L84PfsX7 in 2877 and AT). A total of 386 patients carrying genomic EMD mutations are listed in the Universal Mutation Database on EMD mutations (UMD-EMD; http://www.umd.be/EMD/),3 which was last updated in December 2010. This lists 125 different mutations in 112 probands. With the addition of our data, we take these totals to 130 probands (an increase of 13.8%), 133 different mutations (an increase of 6.0%) and 407 total known mutations (an increase of 5.2%).
The molecular details of the identified mutations are presented in Table 1. The majority (19/21; 90.5%) of the mutations identified here are likely to result in a severely truncated or complete lack of protein. These 19 mutations comprise nine frameshift (9/21; 42.6%), eight non-sense (8/21; 38.1%) and two (2/21; 9.5%) missense mutations (predicted to be M1V but as this is the initiation codon, in practice this will not occur as the mRNA will not be translated). The remaining two mutations were intronic in two unrelated referrals; a carrier female (2370) and a proband (4445). These would be expected to disrupt the highly conserved splice acceptor site at the 3′ end of intron 3 (shown in Figure 1a). We predict that these mutations would deregulate pre-mRNA splicing, although RNA analysis was not conducted. No mutations in the 5′ or 3′ untranslated regions were identified.
The clinical information available on these patients is shown as Supplementary material (Supplementary Table 2). We were unable to obtain a complete clinical data set on all the patients, mainly because each patient was seen in a different clinic. Furthermore these data were collected in 2001 and no follow-up clinical data have been made available to us. Of the 19 patients for whom a family history was available, mutations were mainly of familial origin (15/19; 79%), including one tested in utero (4445) and one as a neonate (2790), with only 21% (4/19) of referrals thought to be of sporadic origin (see Supplementary Table 2). Similarly, the genetic data were also collected in 2001. At this time it was not routine to additionally collect muscle biopsy samples for this disease in these molecular diagnostic laboratories. Therefore there is also an absence of protein analysis data with unknown emerin expression levels.
Eight novel EMD mutations
These eight mutations include six frameshifts mutations (p.D9GfsX24, p.F39SfsX17, p.R45KfsX16, p.F190YfsX19, p.R203PfsX34 and p.R204PfsX7) and two non-sense mutations (p.S143X and p.W200X) (Table 1). Frameshift mutations in the EMD gene, previously shown to allow modified emerin expression, suggest the shortest length required for protein expression is 208 residues,26, 27 which is 94% (208/221) of the nucleoplasmic domain length, as shown by the in vivo expression of the p.P169RfsX40 mutation.28 From this, we can predict that three of our novel mutations (p.F190YfsX19, p.R203PfsX34 and p.R204PfsX7) will express truncated forms of erroneous protein, with the remaining mutations preventing protein expression. As expected, two of these three patients with mutations that are predicted to result in expression of a truncated emerin protein, presented with an indistinguishable phenotype from emerin-null patients. However, the third (2521; p.R203PfsX34) was clinically diagnosed with FSHD before genetic testing, suggesting he may be of a slightly less severe phenotype. An unrelated patient (1196) and carrier (722) both had the novel mutation p.W200X, also predicted to non-code for protein.
EMD mutation spectrum analysis
Comprehensive data analysis on the significance of the EMD mutation spectrum has not been published. To perform such analysis on mutations affecting the introns and coding regions of the EMD gene, we added our 18 proband mutations to the 112 in the UMD-EMD database and analysed all 130 mutations for recurrent patterns. Table 2 categorises the percentage of these 130 EMD mutations by type. Incorporating our new mutations with those listed in the UMD-EMD database, increased the proportion of all mutation types (Table 2), except for the deletion and intronic categories. Although the deletion category was slightly reduced in proportion, it still represents the majority of mutation types. Immunohistochemistry studies show ∼90% of EMD mutations result in no protein expression.29 Table 2 demonstrates ∼80% of these comprise of out of frame deletions (29.2%), insertions (14.6%), non-sense (20.8%) and intronic (14.6%) mutations. The remainder arise from some of the inframe and missense mutations that functionally compromise protein expression.30
We next analysed the 130 proband cohort for mutation hot spots. From the 112 mutations listed in the UMD-EMD database, the top 3 hits cover the following codons: codon 1 (11 hits; 9.8%), codons 34, 51 and 84 (4 hits each; 3.5%) and codons 62, 133 and 226 (3 hits each; 2.7%). Incorporating our data adds two hits to codon 1, corroborating it as a clear hot spot. Nine mutations (including the two we report) in codon 1 prevent it from encoding for methionine, thus inhibiting translation initiation. The remaining four delete the entire coding sequence. There are 12 codons with 2 hits including codon 44 and 171 where we have identified a new hit in each, now taking these to 3 hits. We then examined the mutation distribution by exon. Including our new mutations, there are 111 (94 in UMD-EMD database and 17 reported here) mutations in exons 1–6 and 19 intronic mutations (18 in UMD-EMD database and 1 reported here) in the probands. Table 3 shows the number of observed mutations compared with the expected number per exon, according to relative exon length, and the statistical significance for each exon as calculated by a Chi-squared test. From this we observed that the distribution of EMD mutations is nonrandom. Although exon 6 has similar numbers of expected (37) to observed (36) mutations, exon 2 has 30 observed mutations, against 13 expected. The high χ2 value for exon 2 demonstrates a nearly twofold nonrandom higher relative mutation frequency (267.0 vs 13.8%) than expected. In contrast, exons 4 and 5 have lower than expected number of observed mutations. Exon 2 was first proposed as a mutation hot spot in 2001 when the total number of observed mutations was only 97.31 As this finding has withstood the test of time, it substantiates the evidence that exon 2 is susceptible to mutational events.
We next examined the number of different types of mutations affecting each exon. Exon 2 has the second highest at 17, only coming behind the largest exon, exon 6 with 23 (Table 3). The three most common mutations identified in exon 2 are at codon 51 resulting in p.S52AfsX13 (4 hits; stop at codon 64) and p.Y34X (3 hits). Four of our 18 (22%) probands had mutations in exon 2, of which two are novel but with one also targeting residue S52. The third most commonly hit exon is exon 4, with 12 observed against 17 expected mutations, with 11 different types of mutations.
Discussion
Interest in understanding the aetiology of EDMD has concentrated on the more common autosomal form because of its higher incidence and with the association of the LMNA gene in at least 16 other tissue-specific disorders of muscle, bone and fat, collectively termed the laminopathies.32 Our identification of 18 new X-EDMD patients and three carrier females carrying EMD mutations from a large North American EDMD patient cohort provides a significant contribution to the known EMD mutation spectrum. We show EMD mutation spectrum analysis for the first time.
Approximately 90% of EMD mutations result in the complete absence of protein by immunohistochemical staining. This has led some to propose that the details of individual mutations are irrelevant because the disease mechanism is likely to be similar for all mutations that is, genotype–phenotype correlations are not important in X-EDMD. The few mutations allowing modified emerin protein expression yield emerin protein that is expressed in reduced amounts, which are frequently mislocalised.23, 24, 28, 30, 33, 34 The majority of patients with residual emerin expression display clinical phenotypes indistinguishable from their null counterparts. However, it may be presumptuous to presume that there is no discernable genotype–phenotype correlation, as EMD mutations are so rare, that clinical data collection on EDMD patients is often incomplete because any one clinic sees so few patients.
Our EDMD patient cohort included three patients who were clinically misdiagnosed before genetic testing: two with FSHD (patients 2521 and 6784) and one with Becker muscular dystrophy (patient AT). Again this is consistent with the difficulty of distinguishing EDMD from overlapping phenotypes in the absence of genetic testing.5 This overlapping phenomenon may also explain the clinical heterogeneity of the condition. Indeed, patient AT who was misdiagnosed with Becker muscular dystrophy due to an early age of onset of both the myopathy (18 months) and cardiomyopathy (11 years), may have been an example of a severe form of X-EDMD. Alternatively, his clinical severity may result from digenic inheritance, which has been previously reported in EDMD patients.35 EDMD patient 6784 first presented with facial weakness, consistent with a diagnosis of FSHD. Facial weakness is not normally observed at EDMD disease onset, but can develop subsequently.36 Consistent with digenic inheritance in this patient, an FSHD diagnostic D4Z4 contraction at 4q35 was found.
The unusually high frequency of mutations within exon 2 suggests that either this region of the gene has properties that make it at risk for acquiring mutations or that mutations in this region carry a high risk of generating the EDMD phenotype. An inherent instability such as a tendency towards unequal crossing-over, or chemical predisposition to single nucleotide substitutions may occur at a higher level than average in exon 2 than in the other exons.37 It is also the most GC-rich exon in emerin: 60% constitutes G+C bases, with 80% of the GC bases occurring between c.128-c.173, which is half of the exon. GC-rich regions will on average contain more hypermutable methylated CpG dinculeotides than GC-poor regions, which may contribute to exon 2 being a mutational hot spot.38 Finally, it is interesting to note that two independent groups have reported the existence of emerin transcripts lacking exon 2; GenBank EAW7274339 and GenBank CAI43229.1 (direct sequence submission by P. Heath at Wellcome Trust Sanger Institute, 2009). The functional significance of this second emerin transcript remains to be explored.
References
Emery, A. E. Emery-Dreifuss syndrome. J. Med. Genet. 26, 637–641 (1989).
Emery, A. E. Emery-Dreifuss muscular dystrophy—a 40 year retrospective. Neuromuscul. Disord. 10, 228–232 (2000).
Bonne, G., Yaou, R. B., Beroud, C., Boriani, G., Brown, S., de Visser, M. et al. 108th ENMC International Workshop, 3rd Workshop of the MYO-CLUSTER project: EUROMEN, 7th International Emery-Dreifuss Muscular Dystrophy (EDMD) Workshop, 13-15 September 2002, Naarden, The Netherlands. Neuromuscul. Disord. 13, 508–515 (2003).
Kubo, S., Tsukahara, T., Takemitsu, M., Yoon, K. B., Utsumi, H., Nonaka, I. et al. Presence of emerinopathy in cases of rigid spine syndrome. Neuromuscul. Disord. 8 (1998).
Emery, A. E. The muscular dystrophies. Lancet 359, 687–695 (2002).
Ellis, J. A. Emery-Dreifuss muscular dystrophy at the nuclear envelope: 10 years on. Cell Mol. Life Sci. 63, 2702–2709 (2006).
Emery, A. E. & Dreifuss, F. E. Unusual type of benign x-linked muscular dystrophy. J. Neurol. Neurosurg. Psychiatry. 29, 338–342 (1966).
Yates, J. R., Bagshaw, J., Aksmanovic, V. M., Coomber, E., McMahon, R., Whittaker, J. L. et al. Genotype-phenotype analysis in X-linked Emery-Dreifuss muscular dystrophy and identification of a missense mutation associated with a milder phenotype. Neuromuscul. Disord. 9, 159–165 (1999).
Bione, S., Maestrini, E., Rivella, S., Mancini, M., Regis, S., Romeo, G. et al. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat. Genet. 8, 323–327 (1994).
Gueneau, L., Bertrand, A. T., Jais, J. P., Salih, M. A., Stojkovic, T., Wehnert, M. et al. Mutations of the FHL1 gene cause Emery-Dreifuss muscular dystrophy. Am. J. Hum. Genet. 85, 338–353 (2009).
Norwood, F. L., Harling, C., Chinnery, P. F., Eagle, M., Bushby, K., Straub, V. et al. Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain 132, 3175–3186 (2009).
Nagano, A., Koga, R., Ogawa, M., Kurano, Y., Kawada, J., Okada, R. et al. Emerin deficiency at the nuclear membrane in patients with Emery-Dreifuss muscular dystrophy. Nat. Genet. 12, 254–259 (1996).
Ura, S., Hayashi, Y. K., Goto, K., Astejada, M. N., Murakami, T., Nagato, M. et al. Limb-girdle muscular dystrophy due to emerin gene mutations. Arch. Neurol. 64, 1038–1041 (2007).
Ben Yaou, R., Toutain, A., Arimura, T., Demay, L., Massart, C., Peccate, C. et al. Multitissular involvement in a family with LMNA and EMD mutations: role of digenic mechanism? Neurology 68, 1883–1894 (2007).
Karst, M. L., Herron, K. J. & Olson, T. M. X-linked nonsyndromic sinus node dysfunction and atrial fibrillation caused by emerin mutation. J. Cardiovasc. Electrophysiol. 19, 510–515 (2008).
Schessl, J., Taratuto, A. L., Sewry, C., Battini, R., Chin, S. S., Maiti, B. et al. Clinical, histological and genetic characterization of reducing body myopathy caused by mutations in FHL1. Brain 132, 452–464 (2009).
Quijano-Roy, S., Mbieleu, B., Bonnemann, C. G., Jeannet, P. Y., Colomer, J., Clarke, N. F. et al. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann. Neurol. 64, 177–186 (2008).
Bonne, G., Di Barletta, M. R., Varnous, S., Becane, H. M., Hammouda, E. H., Merlini, L. et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet. 21, 285–288 (1999).
Clements, L., Manilal, S., Love, D. R. & Morris, G. E. Direct interaction between emerin and lamin A. Biochem. Biophys. Res. Commun. 267, 709–714 (2000).
Zhang, Q., Bethmann, C., Worth, N. F., Davies, J. D., Wasner, C., Feuer, A. et al. Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum. Mol. Genet. 16, 2816–2833 (2007).
Brown, C. A., Lanning, R. W., McKinney, K. Q., Salvino, A. R., Cherniske, E., Crowe, C. A. et al. Novel and recurrent mutations in lamin A/C in patients with Emery-Dreifuss muscular dystrophy. Am. J. Med. Genet. 102, 359–367 (2001).
Scharner, J., Brown, C. A., Bower, M., Iannaccone, S. T., Khatri, I. A., Escolar, D. et al. Novel LMNA mutations in patients with Emery-Dreifuss muscular dystrophy and functional characterization of four LMNA mutations. Hum. Mutat. 32, 152–167 (2010).
Ellis, J. A., Yates, J. R., Kendrick-Jones, J. & Brown, C. A. Changes at P183 of emerin weaken its protein-protein interactions resulting in X-linked Emery-Dreifuss muscular dystrophy. Hum. Genet. 104, 262–268 (1999).
Ellis, J. A., Brown, C. A., Tilley, L. D., Kendrick-Jones, J., Spence, J. E. & Yates, J. R. Two distal mutations in the gene encoding emerin have profoundly different effects on emerin protein expression. Neuromuscul. Disord. 10, 24–30 (2000).
van Deutekom, J. C., Wijmenga, C., van Tienhoven, E. A., Gruter, A. M., Hewitt, J. E., Padberg, G. W. et al. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum. Mol. Genet. 2, 2037–2042 (1993).
Tsuchiya, Y., Hase, A., Ogawa, M., Yorifuji, H. & Arahata, K. Distinct regions specify the nuclear membrane targeting of emerin, the responsible protein for Emery-Dreifuss muscular dystrophy. Eur. J. Biochem. 259, 859–865 (1999).
Ostlund, C., Ellenberg, J., Hallberg, E., Lippincott-Schwartz, J. & Worman, H. J. Intracellular trafficking of emerin, the Emery-Dreifuss muscular dystrophy protein. J. Cell Sci. 112 (Part 11), 1709–1719 (1999).
Cartegni, L., di Barletta, M. R., Barresi, R., Squarzoni, S., Sabatelli, P., Maraldi, N. et al. Heart-specific localization of emerin: new insights into Emery-Dreifuss muscular dystrophy. Hum. Mol. Genet. 6, 2257–2264 (1997).
Yates, J. R. & Wehnert, M. The Emery-Dreifuss muscular dystrophy mutation database. Neuromuscul. Disord. 9, 199 (1999).
Ellis, J. A., Craxton, M., Yates, J. R. & Kendrick-Jones, J. Aberrant intracellular targeting and cell cycle-dependent phosphorylation of emerin contribute to the Emery-Dreifuss muscular dystrophy phenotype. J. Cell Sci. 111 (Part 6), 781–792 (1998).
Nevo, Y., Ahituv, S., Yaron, Y., Kedmi, M., Shomrat, R., Legum, C. et al. Novel mutations in the emerin gene in Israeli families. Hum. Mutat. 17, 522 (2001).
Stewart, C. L., Kozlov, S., Fong, L. G. & Young, S. G. Mouse models of the laminopathies. Exp. Cell Res. 313, 2144–2156 (2007).
Fairley, E. A., Kendrick-Jones, J. & Ellis, J. A. The Emery-Dreifuss muscular dystrophy phenotype arises from aberrant targeting and binding of emerin at the inner nuclear membrane. J. Cell Sci. 112 (Part 15), 2571–2582 (1999).
Manilal, S., Recan, D., Sewry, C. A., Hoeltzenbein, M., Llense, S., Leturcq, F. et al. Mutations in Emery-Dreifuss muscular dystrophy and their effects on emerin protein expression. Hum. Mol. Genet. 7, 855–864 (1998).
Muntoni, F., Bonne, G., Goldfarb, L. G., Mercuri, E., Piercy, R. J., Burke, M. et al. Disease severity in dominant Emery Dreifuss is increased by mutations in both emerin and desmin proteins. Brain 129, 1260–1268 (2006).
Pinelli, G., Dominici, P., Merlini, L., Di Pasquale, G., Granata, C. & Bonfiglioli, S. Cardiologic evaluation in a family with Emery-Dreifuss muscular dystrophy. G. Ital. Cardiol. 17, 589–593 (1987).
Schmidt, S., Gerasimova, A., Kondrashov, F. A., Adzhubei, I. A., Kondrashov, A. S. & Sunyaev, S. Hypermutable non-synonymous sites are under stronger negative selection. PLoS Genet. 4, e1000281 (2008).
Subramanian, S. & Kumar, S. Neutral substitutions occur at a faster rate in exons than in noncoding DNA in primate genomes. Genome. Res. 13, 838–844 (2003).
Venter, J. C., Adams, M. D., Myers, E. W., Li, P. W., Mural, R. J., Sutton, G. G et al. The sequence of the human genome. Science 291, 1304–1351 (2001).
Acknowledgements
We wish to thank the curators of the UMD-EMD database, Gisele Bonne and Rabah Ben Yaou for their guidance on how to extract data contained with it. This work was supported by grant No. A6356678101 from the Muscular Dystrophy Association, awarded to JAE and CAB. JS was partially funded by the MYORES Network of Excellence (contract 511978) from the European Commission 6th Framework Programme. The laboratory of PSZ is also supported by The Muscular Dystrophy Campaign, the Association of International Cancer Research, Association Française contre les Myopathies, The Wellcome Trust and OPTISTEM (contract 223098), through the European Union 7th Framework Programme.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Brown, C., Scharner, J., Felice, K. et al. Novel and recurrent EMD mutations in patients with Emery–Dreifuss muscular dystrophy, identify exon 2 as a mutation hot spot. J Hum Genet 56, 589–594 (2011). https://doi.org/10.1038/jhg.2011.65
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2011.65
Keywords
This article is cited by
-
Autosomal dominant Emery-Dreifuss muscular dystrophy caused by a mutation in the lamin A/C gene identified by exome sequencing: a case report
BMC Pediatrics (2022)
-
Targeted next-generation sequencing identified a known EMD mutation in a Chinese patient with Emery-Dreifuss muscular dystrophy
Human Genome Variation (2019)
