
Abstract
22q11.2 duplication syndrome has recently been established as a new syndrome manifesting broad clinical phenotypes including mental retardation. It is reciprocal to DiGeorge (DGS)/velo-cardio-facial syndrome (VCFS), in which the same portion of the chromosome is hemizygously deleted. Deletions and duplications of the 22q11.2 region are facilitated by the low-copy repeats (LCRs) flanking this region. In this study, we aimed to identify the directions of the duplicated segments of 22q11.2 to better understand the mechanism of chromosomal duplication. To achieve this aim, we accumulated samples from four patients with 22q11.2 duplications. One of the patients had an atypically small (741 kb) duplication of 22q11.2. The centromeric end of the breakpoint was on LCR22A, but the telomeric end was between LCR22A and B. Therefore, the duplicated segment did not include T-box 1 gene (TBX1), the gene primarily responsible for the DGS/VCFS. As this duplication was shared by the patient's healthy mother, this appears to be a benign copy-number variation rather than a disease-causing alteration. The other three patients showed 3.0 or 4.0 Mb duplications flanked by LCRs. The directions of the duplicated segments were investigated by fiber-fluorescence in situ hybridization analysis. All samples showed tandem configurations. These results support the hypothesized mechanism of non-allelic homologous recombination with flanking LCRs and add additional evidence that many interstitial duplications are aligned as tandem configurations.
Similar content being viewed by others


Main
In the past decade, a new syndrome, 22q11.2 duplication syndrome, has been identified.1, 2 As the chromosomal segments duplicated in 22q11.2 duplication syndrome overlap those deleted in DiGeorge syndrome (DGS)/velo-cardio-facial syndrome (VCFS), 22q11.2 duplication syndrome is the reciprocal genomic disorder of 22q11.2 deletion syndrome.3 Deletions and duplications of the 22q11.2 region are facilitated by the flanking low-copy repeats (LCR) in this region. There are eight different LCRs located in the 22q11.2 region, identified as LCR22s A–H,4, 5 which mediate non-allelic homologous recombination (NAHR) as the result of unequal crossovers between non-homologous alleles.6 The four LCRs in the proximal region of 22q11.2 seem to be especially prone to mediate NAHR and have been implicated in the rearrangements associated with DGS/VCFS, cat eye syndrome and Emanuel syndrome, all of which are thought to be due to either decreased or increased gene dosage.4, 5
In our study, blood samples were collected from four patients with 22q11.2 duplications and one of their parents (Supplementary Information S1). Genomic copy numbers were analyzed by array comparative genomic hybridization using the Agilent Whole Human Genome Oligo Microarray kit (60 K for patient 1 and 44 K for patients 2–4; Agilent Technologies, Palo Alto, CA, USA), as described elsewhere.7, 8 The only aberrations identified by array comparative genomic hybridization analysis of the four patients were genomic copy number gains at 22q11.2. The range of the duplicated segment in patient 1 was atypically small and was inherited from his healthy mother. No identical duplication could be found in existing databases, including the Database of Genomic Variants, ISCA and DECIPHER. As the location of the distal end of the duplication was between LCR22A and LCR22B (Figure 1), this duplication did not include T-box 1 gene (TBX1), the main disease-causing gene in 22q11.2 deletion syndrome.9 Although some chromosomal duplications show variable phenotypes even in the same family, the phenotypic features of patient 1 are too severe to be explained by clinical heterogeneity.10 Therefore, this 741 kb duplication appears to be a benign copy-number variation and is not related to the severe developmental delay in patient 1.

Physical map of the 22q11.2 region and array comparative genomic hybridization profiles of the patients. Horizontal and vertical axes indicate the physical positions on chromosome 22 and the log2 ratios of the probes, respectively. The blue boxes show the regions of genomic copy number gains, which were determined by the ADM-2 algorithm with a threshold of 6.0. Dots indicate the positions of the probes; blue lines, moving averages; black boxes; LCR22s; Arabic numbers surrounded by red and green boxes, the location and color of the FISH probes. 1, RP11-243A4; 2, CTD-3067F4; 3, CTD-3120F6; 4, RP11-260L7.
Patients 2 and 3 showed a typical 3.0 Mb microduplication of 22q11.2, and patient 4 showed a larger 4.0 Mb duplication flanked by LCRs (Figure 1). The phenotypic appearances of these patients were much milder than that of patient 1. In particular, the chief complaint of patients 3 and 4 was attention deficit/hyperactivity disorder, which is often seen in patients with 22q11.2 duplication syndrome (Supplementary Information S2).11 An increasing number of reports of 3.0 Mb microduplications of 22q11.2 between LCR22A and LCR22D have established this duplication as a clinical entity with highly variable clinical phenotypes, ranging from normal to multiple defects.2 Some features are similar to those often seen in patients with DGS/VCFS, which include congenital heart defects; urogenital abnormalities; velopharyngeal insufficiency with or without cleft palate; and characteristic facial findings, including micrognathia and upslanting palpebral fissures.2, 12, 13, 14 However, there are many patients with 22q11.2 duplications who show only mild neurological abnormalities, including mental retardation, mild developmental delay, autistic features and attention deficit/hyperactivity disorder, as in the present study.11 Therefore, it is difficult to suspect a 22q11.2 duplication on the basis of only the phenotype.
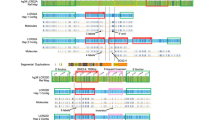
In accordance with the supposed mechanisms of NAHR, duplicated segments are theoretically considered to be inserted in a tandem manner.15 However, experimental evidence of tandem insertions is insufficient. Thus, this study aimed at detecting the directions of the inserted segments by using fiber-fluorescence in situ hybridization (FISH) analysis. Metaphase or interphase chromosomes were prepared and used for FISH analyses with BAC probes selected from an in silico library (UCSC genome browser) confirming the duplications observed in the patients (Figure 2), and then, the directions of the duplicated segments in the patients were analyzed by fiber-FISH analyses on stretched DNA fiber, as previously described,7, 8 and all patients showed tandem configurations regardless of the variable duplication sizes (Figure 3). This result supports the hypothesized mechanism of NAHR in patients 2–4 with 22q11.2 duplications flanked by LCRs. Although the atypically small 741 kb duplication identified in patient 1 and his mother was not flanked by LCRs at the distal end, this result added additional evidence that many interstitial duplications are aligned in tandem configurations.7, 8

Two-color FISH analysis for patient 1. The signal intensity in metaphase of one of the green CTD-3067F4 signals (indicated as ref. 2 in Figure 1) is stronger than that of the other (arrow). Three green signals can be seen in interphase (arrow head). The red signals are from RP11-260L7 (indicated as ref. 4 in Figure 1).

Fiber-FISH analysis showed tandem orientations in all the samples. On the right side, the alignments of the BAC probe signals are indicated by the colored rectangles with Arabic numbers corresponding to those in the physical map (Figure 1).
References
Ensenauer, R. E., Adeyinka, A., Flynn, H. C., Michels, V. V., Lindor, N. M., Dawson, D. B. et al. Microduplication 22q11.2, an emerging syndrome: clinical, cytogenetic, and molecular analysis of thirteen patients. Am. J. Hum. Genet. 73, 1027–1040 (2003).
Portnoï, M.- F. Microduplication 22q11.2: a new chromosomal syndrome. Eur. J. Med. Genet. 52, 88–93 (2009).
Hassed, S. J., Hopcus-Niccum, D., Zhang, L., Li, S. & Mulvihill, J. J. A new genomic duplication syndrome complementary to the velocardiofacial (22q11 deletion) syndrome. Clin. Genet. 65, 400–404 (2004).
Edelmann, L., Pandita, R. K., Spiteri, E., Funke, B., Goldberg, R., Palanisamy, N. et al. A common molecular basis for rearrangement disorders on chromosome 22q11. Hum. Mol. Genet. 8, 1157–1167 (1999).
Shaikh, T. H., Kurahashi, H., Saitta, S. C., O'Hare, A. M., Hu, P., Roe, B. A. et al. Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: genomic organization and deletion endpoint analysis. Hum. Mol. Genet. 9, 489–501 (2000).
Stankiewicz, P. & Lupski, J. R. Genome architecture, rearrangements and genomic disorders. Trends Genet. 18, 74–82 (2002).
Shimojima, K., Tanaka, K. & Yamamoto, T. A de novo intra-chromosomal tandem duplication at 22q13.1q13.31 including the Rubinstein-Taybi region but with no bipolar disorder. Am. J. Med. Genet. A 149A, 1359–1363 (2009).
Shimojima, K., Imai, K. & Yamamoto, T. A de novo 22q11.22q11.23 interchromosomal tandem duplication in a boy with developmental delay, hyperactivity, and epilepsy. Am. J. Med. Genet. A. 152A, 2820–2826 (2010).
Yagi, H., Furutani, Y., Hamada, H., Sasaki, T., Asakawa, S., Minoshima, S. et al. Role of TBX1 in human del22q11.2 syndrome. Lancet. 362, 1366–1373 (2003).
Ou, Z., Berg, J. S., Yonath, H., Enciso, V. B., Miller, D. T., Picker, J. et al. Microduplications of 22q11.2 are frequently inherited and are associated with variable phenotypes. Genet. Med. 10, 267–277 (2008).
Courtens, W., Schramme, I. & Laridon, A. Microduplication 22q11.2: a benign polymorphism or a syndrome with a very large clinical variability and reduced penetrance?--Report of two families. Am. J. Med. Genet. A. 146A, 758–763 (2008).
Schramm, C., Draaken, M., Bartels, E., Boemers, T. M., Aretz, S., Brockschmidt, F. F. et al. De novo microduplication at 22q11.21 in a patient with VACTERL association. Eur. J. Med. Genet. 54, 9–13 (2011).
Lundin, J., Soderhall, C., Lunden, L., Hammarsjo, A., White, I., Schoumans, J. et al. 22q11.2 microduplication in two patients with bladder exstrophy and hearing impairment. Eur. J. Med. Genet. 53, 61–65 (2010).
Draaken, M., Reutter, H., Schramm, C., Bartels, E., Boemers, T. M., Ebert, A. K. et al. Microduplications at 22q11.21 are associated with non-syndromic classic bladder exstrophy. Eur. J. Med. Genet. 53, 55–60 (2010).
Gu, W., Zhang, F. & Lupski, J. R. Mechanisms for human genomic rearrangements. Pathogenetics. 1, 4 (2008).
Acknowledgements
This work was partially supported by a grant from the Hayashi Memorial Foundation for Female Natural Scientists (KS) and a research grant, Scientific Research (c), 21591334, 2010 (TY), from the Japan Ministry of Education, Science, Sports and Culture.
Author information
Authors and Affiliations
Corresponding author
Additional information
WEB-BASED DATABASE
The Database of Genomic Variants: http://projects.tcag.ca/variation/
The International Standards For Cytogenomic Arrays Consortium (ISCA): https://www.iscaconsortium.org/
DECIPHER: http://decipher.sanger.ac.uk/
UCSC Human genome browser: http://genome.ucsc.edu/
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Shimojima, K., Okamoto, N., Inazu, T. et al. Tandem configurations of variably duplicated segments of 22q11.2 confirmed by fiber-FISH analysis. J Hum Genet 56, 810–812 (2011). https://doi.org/10.1038/jhg.2011.100
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2011.100
