
Abstract
This report describes androgenetic/biparental mosaicism in a 4-year-old Japanese girl with Beckwith–Wiedemann syndrome (BWS)-like and paternal uniparental disomy 14 (upd(14)pat)-like phenotypes. We performed methylation analysis for 18 differentially methylated regions on various chromosomes, genome-wide microsatellite analysis for a total of 90 loci and expression analysis of SNRPN in leukocytes. Consequently, she was found to have an androgenetic 46,XX cell lineage and a normal 46,XX cell lineage, with the frequency of the androgenetic cells being roughly calculated as 91% in leukocytes, 70% in tongue tissues and 79% in tonsil tissues. It is likely that, after a normal fertilization between an ovum and a sperm, the paternally derived pronucleus alone, but not the maternally derived pronucleus, underwent a mitotic division, resulting both in the generation of the androgenetic cell lineage by endoreplication of one blastomere containing a paternally derived pronucleus and in the formation of the normal cell lineage by union of paternally and maternally derived pronuclei. It appears that the extent of overall (epi)genetic aberrations exceeded the threshold level for the development of BWS-like and upd(14)pat-like phenotypes, but not for the occurrence of other imprinting disorders or recessive Mendelian disorders.
Similar content being viewed by others


Introduction
A pure androgenetic human with paternal uniparental disomy for all chromosomes is incompatible with life because of genomic imprinting.1, 2 However, a human with an androgenetic cell lineage could be viable in the presence of a normal cell lineage. Indeed, an androgenetic cell lineage has been identified in six liveborn individuals with variable phenotypes.3, 4, 5, 6, 7 All the androgenetic cell lineages have a 46,XX karyotype, and this is consistent with the lethality of an androgenetic 46,YY cell lineage.
Here, we report on a girl with androgenetic/biparental mosaicism, and discuss the underlying factors for the phenotypic development.
Case report
This patient was conceived naturally to non-consanguineous and healthy parents. At 24 weeks gestation, the mother was referred to us because of threatened premature delivery. Ultrasound studies showed Beckwith–Wiedemann syndrome (BWS)-like features,8 such as macroglossia, organomegaly and umbilical hernia, together with polyhydramnios and placentomegaly. The mother repeatedly received amnioreduction and tocolysis.
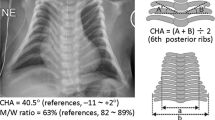
She was delivered by an emergency cesarean section because of preterm rupture of membranes at 34 weeks of gestation. Her birth weight was 3730 g (+4.8 s.d. for gestational age), and her length 45.6 cm (+0.7 s.d.). The placenta weighed 1040 g (+7.3 s.d.).9 She was admitted to a neonatal intensive care unit due to asphyxia. Physical examination confirmed a BWS-like phenotype. Notably, chest roentgenograms delineated mild bell-shaped thorax characteristic of paternal uniparental disomy 14 (upd(14)pat),10 although coat hanger appearance of the ribs indicative of upd(14)pat was absent (Supplementary Figure 1). She was placed on mechanical ventilation for 2 months, and received tracheostomy, glossectomy and tonsillectomy in her infancy, due to upper airway obstruction. She also had several clinical features occasionally reported in BWS8 (Supplementary Table 1). Her karyotype was 46,XX in all the 50 lymphocytes analyzed. On the last examination at 4 years of age, she showed postnatal growth failure and severe developmental retardation.
Molecular studies
This study was approved by the Institutional Review Board Committee at the National Center for Child health and Development, and performed after obtaining informed consent.
Methylation analysis
We first performed bisulfite sequencing for the H19-DMR (differentially methylated region) and KvDMR1 as a screening of BWS11, 12 and that for the IG-DMR and the MEG3-DMR as a screening of upd(14)pat,10 using leukocyte genomic DNA. Paternally derived clones were predominantly identified for the four DMRs examined (Figure 1a). We next performed combined bisulfite restriction analysis for multiple DMRs, as reported previously.13 All the autosomal DMRs exhibited markedly skewed methylation patterns consistent with predominance of paternally inherited clones, whereas the XIST-DMR on the X chromosome showed a normal methylation pattern (Figure 1a).

Representative molecular results. (a) Methylation analysis. Upper part: Bisulfite sequencing data for the H19-DMR and the KvDMR1 on 11p15.5, and those for the IG-DMR and the MEG3-DMR on 14q32.2. Each line indicates a single clone, and each circle denotes a CpG dinucleotide; filled and open circles represent methylated and unmethylated cytosines, respectively. Paternally expressed genes are shown in blue, maternally expressed gene in red, and the DMRs in green. The H19-DMR, the IG-DMR, and the MEG3-DMR are usually methylated after paternal transmission and unmethylated after maternal transmission, whereas the KvDMR1 is usually unmethylated after paternal transmission and methylated after maternal transmission.10, 11 Lower part: Methylation indices (the ratios of methylated clones) obtained from the COBRA analyses for the 18 DMRs. The DMRs highlighted in blue and pink are methylated after paternal and maternal transmissions, respectively. The black vertical bars indicate the reference data (maximum – minimum) in leukocyte genomic DNA of 20 normal control subjects (the XIST-DMR data are obtained from 16 control females). (b) Representative microsatellite analysis. Major peaks of paternal origin and minor peaks of maternal origin (red arrows) have been identified in this patient. The minor peaks of maternal origin are more obvious in tongue and tonsil tissues than in leukocytes (Leu.). (c) Relative expression level (mean±s.d.) of SNRPN. The data are normalized against TBP. SRS: an SRS patient with an epimutation (hypomethylation) of the H19-DMR; BWS1: a BWS patient with an epimutation (hypermethylation) of the H19-DMR; BWS2: a BWS patient with upd(11)pat; PWS1: a Prader-Willi syndrome (PWS) patient with upd(15)mat; PWS2: a PWS patient with an epimutation (hypermethylation) of the SNRPN-DMR; AS1: an Angelman syndrome (AS) patient with upd(15)pat; and AS2: an AS patient with an epimutation (hypomethylation) of the SNRPN-DMR. The data were obtained using an ABI Prism 7000 Sequence Detection System (Applied Biosystems).
Genome-wide microsatellite analysis
Microsatellite analysis was performed for 90 loci with high heterozygosities in the Japanese population.14 Major peaks consistent with paternal uniparental isodisomy and minor peaks of maternal origin were identified for at least one locus on each chromosome, with the minor peaks of maternal origin being more obvious in tongue and tonsil tissues than in leukocytes (Figure 1b and Supplementary Table 2). There were no loci with three or four peaks indicative of chimerism. The frequency of the androgenetic cells was calculated as 91% in leukocytes, 70% in tongue cells and 79% in tonsil cells, although the estimation apparently was a rough one (for details, see Supplementary Methods).
Expression analysis
We examined SNRPN expression, because SNRPN showed strong expression in leukocytes (for details, see Supplementary Data). SNRPN expression was almost doubled in the leukocytes of this patient (Figure 1c).
Discussion
These results suggest that this patient had an androgenetic 46,XX cell lineage and a normal 46,XX cell lineage. In this regard, both the androgenetic and the biparental cell lineages appear to have derived from a single sperm and a single ovum, because a single haploid genome of paternal origin and that of maternal origin were identified in this patient by genome-wide microsatellite analysis. Thus, it is likely that after a normal fertilization between an ovum and a sperm, the paternally derived pronucleus alone, but not the maternally derived pronucleus, underwent a mitotic division, resulting both in the generation of the androgenetic cell lineage by endoreplication of one blastomere containing a paternally derived pronucleus and in the formation of the normal cell lineage by union of paternally and maternally derived pronuclei (Figure 2). This model has been proposed for androgenetic/biparental mosaicism generated after fertilization between a single ovum and a single sperm.5, 15, 16 The normal methylation pattern of the XIST-DMR is explained by assuming that the two X chromosomes in the androgenetic cell lineage undergo random X-inactivation, as in the normal cell lineage. Furthermore, the results of microsatellite analysis imply that the androgenetic cells were more prevalent in leukocytes than in tongue and tonsil tissues.

Schematic representation of the generation of the androgenetic/biparental mosaicism. Polar bodies are not shown.
A somatic androgenetic cell lineage has been identified in seven liveborn patients including this patient (Supplementary Table 1).3, 4, 5, 6, 7 In this context, leukocytes are preferentially utilized for genetic analyses in human patients, and detailed examinations such as analyses of plural DMRs are necessary to detect an androgenetic cell lineage. Thus, the hitherto identified patients would be limited to those who had androgenetic cells as a predominant cell lineage in leukocytes probably because of a stochastic event and received detailed molecular studies. If so, an androgenetic cell lineage may not be so rare, and could be revealed by detailed analyses as well as examinations of additional tissues in patients with relatively complex phenotypes, as observed in the present patient.
Phenotypic features in androgenetic/biparental mosaicism would be determined by several factors. They include (1) the ratio of two cell lineages in various tissues/organs, (2) the number of imprinted domains relevant to specific features (for example, dysregulation of the imprinted domains on 11p15.5 and 14q32.2 is involved in placentomegaly9, 17), (3) the degree of clinical effects of dysregulated imprinted domains (an (epi)dominant effect has been assumed for the 11p15.5 imprinted domains18), (4) expression levels of imprinted genes in androgenetic cells (although SNRPN expression of this patient was consistent with androgenetic cells being predominant in leukocytes, complicated expression patterns have been identified for several imprinted genes in both androgenetic and parthenogenetic fetal mice, probably because of perturbed cis- and trans-acting regulatory mechanisms19) and (5) unmasking of possible paternally inherited recessive mutation(s) in androgenetic cells. Thus, in this patient, it appears that the extent of overall (epi)genetic aberrations exceeded the threshold level for the development of BWS-like and upd(14)pat-like body and placental phenotypes, but remained below the threshold level for the occurrence of other imprinting disorders or recessive Mendelian disorders.
References
Surani, M. A., Barton, S. C. & Norris, M. L. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature 308, 548–550 (1984).
McGrath, J. & Solter, D. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell 37, 179–183 (1984).
Hoban, P. R., Heighway, J., White, G. R., Baker, B., Gardner, J., Birch, J. M. et al. Genome-wide loss of maternal alleles in a nephrogenic rest and Wilms’ tumour from a BWS patient. Hum. Genet. 95, 651–656 (1995).
Bryke, C. R., Garber, A. T. & Israel, J. Evolution of a complex phenotype in a unique patient with a paternal uniparental disomy for every chromosome cell line and a normal biparental inheritance cell line. Am. J. Hum. Genet. 75 (Suppl), 831 (2004).
Giurgea, I., Sanlaville, D., Fournet, J. C., Sempoux, C., Bellanne-Chantelot, C. & Touati, G. Congenital hyperinsulinism and mosaic abnormalities of the ploidy. J. Med. Genet. 43, 248–254 (2006).
Wilson, M., Peters, G., Bennetts, B., McGillivray, G., Wu, Z. H., Poon, C. et al. The clinical phenotype of mosaicism for genome-wide paternal uniparental disomy: two new reports. Am. J. Med. Genet. Part A 146A, 137–148 (2008).
Reed, R. C., Beischel, L., Schoof, J., Johnson, J., Raff, M. L. & Kapur, R. P. Androgenetic/biparental mosaicism in an infant with hepatic mesenchymal hamartoma and placental mesenchymal dysplasia. Pediatr. Dev. Pathol. 11, 377–383 (2008).
Jones, K. L. Smith's Recognizable Patterns of Human Malformation 6th edn. (Elsevier Saunders: Philadelphia, 2006).
Kagami, M., Yamazawa, K., Matsubara, K., Matsuo, N. & Ogata, T. Placentomegaly in paternal uniparental disomy for human chromosome 14. Placenta 29, 760–761 (2008).
Kagami, M., Sekita, Y., Nishimura, G., Irie, M., Kato, F., Okada, M. et al. Deletions and epimutations affecting the human 14q32.2 imprinted region in individuals with paternal and maternal upd(14)-like phenotypes. Nat. Genet. 40, 237–242 (2008).
Yamazawa, K., Kagami, M., Nagai, T., Kondoh, T., Onigata, K., Maeyama, K. et al. Molecular and clinical findings and their correlations in Silver-Russell syndrome: implications for a positive role of IGF2 in growth determination and differential imprinting regulation of the IGF2-H19 domain in bodies and placentas. J. Mol. Med. 86, 1171–1181 (2008).
Weksberg, R., Shuman, C. & Beckwith, J. B. Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 18, 8–14 (2010).
Yamazawa, K., Nakabayashi, K., Kagami, M., Sato, T., Saitoh, S., Horikawa, R. et al. Parthenogenetic chimaerism/mosaicism with a Silver-Russell syndrome-like phenotype. J. Med. Genet. 47, 782–785 (2010).
Ikari, K., Onda, H., Furushima, K., Maeda, S., Harata, S. & Takeda, J. Establishment of an optimized set of 406 microsatellite markers covering the whole genome for the Japanese population. J. Hum. Genet. 46, 207–210 (2001).
Kaiser-Rogers, K. A., McFadden, D. E., Livasy, C. A., Dansereau, J., Jiang, R., Knops, J. F. et al. Androgenetic/biparental mosaicism causes placental mesenchymal dysplasia. J. Med. Genet. 43, 187–192 (2006).
Kotzot, D. Complex and segmental uniparental disomy updated. J. Med. Genet. 45, 545–556 (2008).
Monk, D., Arnaud, P., Apostolidou, S., Hills, F. A., Kelsey, G., Stanier, P. et al. Limited evolutionary conservation of imprinting in the human placenta. Proc. Natl. Acad. Sci. USA. 103, 6623–6628 (2006).
Azzi, S., Rossignol, S., Steunou, V., Sas, T., Thibaud, N., Danton, F. et al. Multilocus methylation analysis in a large cohort of 11p15-related foetal growth disorders (Russell Silver and Beckwith Wiedemann syndromes) reveals simultaneous loss of methylation at paternal and maternal imprinted loci. Hum. Mol. Genet. 18, 4724–4733 (2009).
Ogawa, H., Wu, Q., Komiyama, J., Obata, Y. & Kono, T. Disruption of parental-specific expression of imprinted genes in uniparental fetuses. FEBS Lett. 580, 5377–5384 (2006).
Acknowledgements
This work was supported by grants from the Ministry of Health, Labor, and Welfare, and the Ministry of Education, Science, Sports and Culture.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Yamazawa, K., Nakabayashi, K., Matsuoka, K. et al. Androgenetic/biparental mosaicism in a girl with Beckwith–Wiedemann syndrome-like and upd(14)pat-like phenotypes. J Hum Genet 56, 91–93 (2011). https://doi.org/10.1038/jhg.2010.142
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2010.142
Keywords
This article is cited by
-
Parthenogenetic mosaicism: generation via second polar body retention and unmasking of a likely causative PER2 variant for hypersomnia
Clinical Epigenetics (2021)
-
Genome-wide uniparental diploidy of all paternal chromosomes in an 11-year-old girl with deafness and without malignancy
Journal of Human Genetics (2018)
-
Kagami–Ogata syndrome: a clinically recognizable upd(14)pat and related disorder affecting the chromosome 14q32.2 imprinted region
Journal of Human Genetics (2016)
-
Genome-wide paternal uniparental disomy mosaicism in a woman with Beckwith–Wiedemann syndrome and ovarian steroid cell tumour
European Journal of Human Genetics (2013)
-
Epigenetic and genetic alterations of the imprinting disorder Beckwith–Wiedemann syndrome and related disorders
Journal of Human Genetics (2013)
