
Abstract
Mutations in ROR2, encoding the receptor tyrosine kinase-like orphan receptor 2, cause two distinct skeletal diseases: autosomal dominant brachydactyly type B1 (BDB1) and autosomal recessive Robinow syndrome. In a large Chinese family with a limb phenotype, consisting of atypical BDB1 and cutaneous syndactyly of varying degrees, we performed a two-point linkage analysis using microsatellite markers on 2q33–q37 and 9q22.31, and found a significant linkage to the ROR2 locus. We identified a novel single-base deletion in ROR2, c.2243delC (p.W749fsX24), and confirmed its segregation with the limb phenotype in the family. This deletion is predicted to produce a truncated ROR2 protein with an additional C-terminal polypeptide of 24 amino-acid residues. To the best of our knowledge, the deletion represents the second ROR2 mutation associated with a BDB1-syndactyly phenotype.
Similar content being viewed by others


Introduction
The human ROR2 gene, encoding the receptor tyrosine kinase-like orphan receptor 2, maps to chromosome 9q22.31, contains nine exons and spans a genomic length of approximately 228 kb. Mutations in this gene can cause two distinct allelic skeletal diseases.1 Heterozygous gain-of-function mutations in ROR2 are responsible for the autosomal dominant brachydactyly type B1 (BDB1; MIM 113000), whereas homozygous loss-of-function mutations in ROR2 lead to autosomal recessive Robinow syndrome (MIM 268310).2, 3, 4, 5 BDB1 is the most severe type of human brachydactylies, and shows high penetrance and variable expression. Hypoplastic or absent distal phalanges and nails of digits 2–5 in the hands and feet are cardinal phenotypic features of BDB1. The middle phalanges of digits 2–5 are usually short and may form a bony fusion with the corresponding hypoplastic distal phalanges. The deformed thumbs are often flat, broad or bifid. A rarer feature of BDB1 is cutaneous syndactyly affecting both fingers and toes.2 In contrast, Robinow syndrome presents with short stature, short limbs, brachydactyly, hemivertebrae, rib fusion, genital hypoplasia and a characteristic facial appearance.4, 5
Many ROR2 mutations have been reported in patients with BDB1 and the autosomal recessive form of Robinow syndrome.1, 2, 3, 4, 5, 6, 7, 8 However, only the c.2249delG (p.G750fsX23) mutation was found to cause atypical and severe BDB1 associated with cutaneous syndactyly.2 Here, we report a new single-base deletion of the ROR2 gene in a large Chinese family with the BDB1-syndactyly phenotype.
Materials and methods
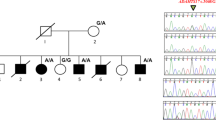
We ascertained a large Chinese family with atypical BDB1 and cutaneous syndactyly of varying degree. The family had 12 affected individuals in three generations (Figure 1a) and 10 of them were said to have “shortened and webbed fingers and toes” by their family members. Five affected individuals were physically examined, and digital photographs were taken. Radiographs of the hands and feet were taken for the proband and her father (III-6 and II-6) (Figure 1a). The proband showed hypoplastic distal phalanges of digits 2–5 of the hands and feet and marked hypoplasia of the third toes (III-6, Figure 1b). All the other four affected individuals had bilateral syndactyly, more marked hypoplastic distal phalanges and absent nails of fingers 2 and 5, and flat and broad thumbs (Figure 1b). Among them, three displayed cutaneous syndactyly of fingers 3–4, two of which also had syndactyly of toes 2–3 or 2–4 (II-6 and II-14, Figure 1b), and one exhibited cutaneous syndactyly of fingers 2–4 and toes 2–3 or 2–4 (II-2, Figure 1b), closely resembling the syndactyly pattern described earlier in the Portuguese family with the c.2249delG mutation of ROR2.2 On X-ray examination, the proband was found to have bifid distal phalanges of the thumbs, hypoplastic distal phalanges of fingers 2–4, with distal symphalangism of fingers 2 and 3, absent distal phalanges of finger 5 and biphalangeal toes 2–5 (III-6 in Figure 1c). In addition to the biphalangeal toes 2–5, the father showed bifid distal and broad proximal phalanges of the thumbs with symphalangism, less marked hypoplastic distal phalanges of the syndactylous fingers 3–4 and absent distal phalanges of finger 5, with more marked hypoplastic middle phalanges (II-6 in Figure 1c).

Pedigree and photographs. (a) Pedigree showing autosomal dominant inheritance. Filled symbols represent affected individuals having BDB1 with or without cutaneous syndactyly, and open symbols represent individuals with a normal limb phenotype. Circles and squares indicate female and male individuals, respectively. Individuals with blood samples collected are marked with an asterisk. The proband (III-6) is indicated by an arrow. (b) Photographs showing limb phenotypes. Examples show typical BDB1 in III-6, severe BDB1 with cutaneous syndactyly of fingers 3–4 and toes 2–4 in II-6 and of fingers 3–4 and toes 2–3 in II-14, and severe BDB1 with cutaneous syndactyly of fingers 2–4 and toes 2–3 (right) or toes 2–4 (left) in II-2. (c) Radiographs of the hands and feet in the proband (III-6) and her father (II-6).
We collected blood samples and extracted genomic DNA from 14 family members, including six affected individuals (Figure 1a), with informed consent from all participating family members. For linkage and haplotype analysis, we selected 14 polymorphic microsatellite markers, including 11 from chromosome 2q33–q37 (D2S2979, D2S2322, D2S334, D2S1345, D2S2382, D2S301, D2S1323, D2S279, D2S1363, D2S345 and D2S140) and 3 from chromosome 9q22.31 (D9S1815, D9S1781 and D9S1841). We carried out two-point linkage analysis as described earlier.9 For mutation detection, we PCR-amplified ROR2 exons 8 and 9 and their flanking intronic sequences using the primer pairs ROR2E8F (5′-GGTTGGTAGAGAACTTAGAGT-3′)/ROR2E8R (5′-ATAATTATGTGCTATGTATCAAG-3′) and ROR2E9F (5′-TAACTAGAGAGCTGTGGGTG-3′)/ROR2E9R (5′-GCTGAGTATGGTGTCTTCTC-3′). The PCR products were then subjected to automatic sequencing after purification. To detect the c.2243delC mutation, the PCR amplicons generated with primers ROR2delF (5′-TCTTCAGCTACGGCCTGCAG-3′) and ROR2delR (5′-CCCGAGGTCTGCGCCGAACT-3′) were digested with restriction enzyme AluI and separated by agarose gel electrophoresis. All available family members and 108 unrelated normal controls were included in the AluI restriction analysis.
Results and discussion
The above-mentioned atypical BDB1 family also displayed a syndactyly phenotype mimicking syndactyly type I (SD1; MIM 185900), which has been mapped to chromosome 2q24–q36.10, 11 Therefore, we first typed genetic markers on 2q33–q37 and 9q22.31 to map the disease locus. Our two-point linkage analysis yielded a maximum logarithmic odds score of 2.71 at θ=0 for markers D9S1815 and D9S1841, supporting a significant genetic linkage of the limb phenotype to the ROR2 locus on 9q22.31 (see Supplementary Table 1). We then screened the last two ROR2 exons for pathogenic mutations in the proband. We identified a heterozygous single-base deletion, c.2243delC, in ROR2 exon 9 (Figure 2a). This deletion created an AluI restriction site in the mutant allele. Using primers ROR2delF and ROR2delR, a PCR fragment of 235 bp was first produced and then digested with the AluI enzyme. The fragment can be cut into two fragments (190 and 45 bp) in the mutant but not in the wild type. By restriction analysis using the AluI enzyme, we confirmed the mutation in all available affected individuals of the family but did not detect the mutation in unaffected family members (Figure 2b) or in 108 unrelated normal controls. The c.2243delC mutation, also designated p.W749fsX24 at the protein level, is predicted to result in a frameshift, producing a truncated ROR2 protein with an additional C-terminal polypeptide of 24 amino-acid residues (Figure 2c). Taken together, our results strongly support a causal relationship between the c.2243delC (p.W749fsX24) mutation in ROR2 and the BDB1-syndactyly phenotype in this large Chinese family.

Identification of the c.2243delC ROR2 mutation. (a) Sequencing chromatogram showing the heterozygous c.2243delC mutation in ROR2 (right) and the wild type (left). (b) AluI restriction analysis showing the segregation of the c.2243delC mutation, shown as a 190-bp fragment, with the limb phenotype in the family. M, DNA marker. (c) Schematic diagram of the wild-type and mutant ROR2 proteins showing different domains and the novel C-terminal peptides. Ig: immunoglobulin-like domain; CRD: frizzled-like cysteine-rich domain; Kr: kringle domain; TM: transmembrane region; TK: tyrosine kinase domain; S/T: serine/threonine-rich domain; PRD: proline-rich domain. Amino-acid residues of the novel C-terminal peptides in the G750fsX23 and W749fsX24 mutants are given with the 23 identical residues in black.
To date, eight different pathogenic ROR2 mutations have been reported in a total of 14 families with BDB1.1, 2, 3, 6, 7, 8 These mutations are believed to have a specific gain-of-function effect, not a simple haploinsufficiency.1, 3 All the documented BDB1-causing ROR2 mutations are nonsense or frameshift and are located in the last two exons or in the last intron,1, 2, 3, 6, 7, 8 enabling the mutant mRNAs to escape degradation by nonsense-mediated decay.12 The five distal mutations clustered between the intracellular tyrosine kinase domain and the first serine/threonine-rich domain cause a more severe phenotype.2, 3, 6, 8 It is worth noting that the three distal ROR2 mutations, c.2246G>A (p.W749X), c.2247G>A (p.W749X) and c.2249delG (p.G750fsX23), have been shown to be associated with BDB1 phenotypes of different severity.2, 6 Whereas the W749X-truncated ROR2 protein has been linked to typical BDB1,2, 6 the G750fsX23 mutant has been documented in the Portuguese family, with both of the two affected individuals showing an atypical BDB1 phenotype with cutaneous syndactyly of digits 2–4.2 Interestingly, the G750fsX23 mutant ROR2 protein has an additional C-terminal polypeptide of 23 amino-acid residues that are identical to the last 23 residues of the W749fsX24 mutant described in this study (Figure 2c). Given the fact that the c.2243delC (p.W749fsX24) and c.2249delG (p.G750fsX23) mutations lead to a very similar BDB1-syndactyly phenotype and produce two mutant ROR2 proteins with identical extra polypeptides at their C-termini, it is conceivable that the additional C-terminal peptide in the mutant ROR2 proteins might contribute to the syndactylous phenotype.
Unlike brachydactyly type B2 (BDB2; MIM 611377) caused by mutations in NOG,13 cutaneous syndactyly has been less frequently observed as an additional phenotypic feature in BDB1. In four different BDB1 families with the c.2265C>A (p.Y755X) ROR2 mutation, only one of the 24 examined patients was reported to have unilateral cutaneous syndactyly of fingers 3–4.2, 8 We also examined 25 affected individuals in a large Chinese BDB1 family with the c.2246G>A (p.W749X) ROR2 mutation, and found one with unilateral cutaneous syndactyly of fingers 3–4 and one with bilateral cutaneous syndactyly of toes 2–3 (data not shown). In marked contrast, 10 of the 12 affected individuals in the Chinese family described in this study showed cutaneous syndactyly. Together, these observations strongly support the possibility of a genotype–phenotype correlation in BDB1 with or without additional syndactyly.
References
Afzal, A. R. & Jeffery, S. One gene, two phenotypes: ROR2 mutations in autosomal recessive Robinow syndrome and autosomal dominant brachydactyly type B. Hum. Mutat. 22, 1–11 (2003).
Oldridge, M., Fortuna, A. M., Maringa, M., Propping, P., Mansour, S., Pollitt, C. et al. Dominant mutations in ROR2, encoding an orphan receptor tyrosine kinase, cause brachydactyly type B. Nat. Genet. 24, 275–278 (2000).
Schwabe, G. C., Tinschert, S., Buschow, C., Meinecke, P., Wolff, G., Gillessen-Kaesbach, G. et al. Distinct mutations in the receptor tyrosine kinase gene ROR2 cause brachydactyly type B. Am. J. Hum. Genet. 67, 822–831 (2000).
Afzal, A. R., Rajab, A., Fenske, C. D., Oldridge, M., Elanko, N., Ternes-Pereira, E. et al. Recessive Robinow syndrome, allelic to dominant brachydactyly type B, is caused by mutation of ROR2. Nat. Genet. 25, 419–422 (2000).
van Bokhoven, H., Celli, J., Kayserili, H., van Beusekom, E., Balci, S., Brussel, W. et al. Mutation of the gene encoding the ROR2 tyrosine kinase causes autosomal recessive Robinow syndrome. Nat. Genet. 25, 423–426 (2000).
Bacchelli, C., Wilson, L., Cook, J., Winter, R. & Goodman, F. ROR2 is mutated in hereditary brachydactyly with nail dysplasia, but not in Sorsby syndrome. Clin. Genet. 64, 263–265 (2003).
Yang, W., Tan, F. Q., Sun, M., Zeng, X., Liu, J., Liu, G. Y. et al. Identification of a recurrent mutation in the ROR2 gene in a Chinese family with brachydactyly type B. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 21, 61–63 (2004).
Hamamy, H., Saleh, N., Oldridge, M., Al-Hadidy, A. & Ajlouni, K. Brachydactyly type B1: report of a family with de novo ROR2 mutation. Clin. Genet. 70, 538–540 (2006).
Zhao, X., Sun, M., Zhao, J., Leyva, J. A., Zhu, H., Yang, W. et al. Mutations in HOXD13 underlie syndactyly type V and a novel brachydactyly-syndactyly syndrome. Am. J. Hum. Genet. 80, 361–371 (2007).
Bosse, K., Betz, R. C., Lee, Y. A., Wienker, T. F., Reis, A., Kleen, H. et al. Localization of a gene for syndactyly type 1 to chromosome 2q34–q36. Am. J. Hum. Genet. 67, 492–497 (2000).
Ghadami, M., Majidzadeh-A, K., Haerian, B. S., Damavandi, E., Yamada, K., Pasallar, P. et al. Confirmation of genetic homogeneity of syndactyly type 1 in an Iranian family. Am. J. Med. Genet. 104, 147–151 (2001).
Ben-Shachar, S., Khajavi, M., Withers, M., Shaw, C., van Bokhoven, H., Brunner, H. et al. Dominant versus recessive traits conveyed by allelic mutations—to what extent is nonsense-mediated decay involved? Clin. Genet. 75, 394–400 (2009).
Lehmann, K., Seemann, P., Silan, F, Goecke, T. O., Irgang, S., Kjaer, K. W. et al. A new subtype of brachydactyly type B caused by point mutations in the bone morphogenetic protein antagonist NOGGIN. Am. J. Hum. Genet. 81, 388–396 (2007).
Acknowledgements
We would like to thank all the family members for their participation in this study. This work was mainly supported by the National Natural Science Foundation of China (30730097 and 30721063) and the Ministry of Science and Technology of China (2005DKA21302) (to XZ). XZ is a Chang Jiang Scholar of Genetic Medicine supported by the Ministry of Education of China.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website (http://www.nature.com/jhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Lv, D., Luo, Y., Yang, W. et al. A novel single-base deletion in ROR2 causes atypical brachydactyly type B1 with cutaneous syndactyly in a large Chinese family. J Hum Genet 54, 422–425 (2009). https://doi.org/10.1038/jhg.2009.48
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2009.48
Keywords
This article is cited by
-
A novel variant in the ROR2 gene underlying brachydactyly type B: a case report
BMC Pediatrics (2022)
