
Abstract
Height is a complex genetic trait that involves multiple genetic loci. Recently, 44 loci associated with height were identified in Caucasian individuals by large-scale genome-wide association (GWA) studies. To identify genetic variants influencing height in the Korean population, we analyzed GWA data from 8842 Korean individuals and identified 15 genomic regions with one or more sequence variants associated with height (P<1 × 10−5). Of these, eight loci were newly identified in Koreans (SUPT3H, EXT1, FREM1, PALM2-AKAP2, NUP37-PMCH, IGF1, KRT20 and ANKRD60). The 15 significant loci account for approximately 1.0% of height variation, with a 3.7-cm difference between individuals with ⩽8 height-increasing alleles (5.1%) and ⩾19 height-increasing alleles (4.2%). We also examined the associations between height loci and idiopathic short stature (ISS). Five loci (SPAG17, KBTBD8, HHIP, HIST1H1D and ACAN) were significantly associated with ISS (uncorrected P<0.05), indicating that height-associated genes in the adult population are involved in extreme cases of short stature in children. This study validates previous reports of loci associated with human height and identified novel candidate regions involved in human growth and development.
Similar content being viewed by others

Introduction
Adult height, a complex genetic trait that involves multiple genetic loci, is a suitable phenotypic trait for genetic studies owing to the high heritability (h2 ∼0.75 to 0.9) and the relatively limited contribution of environmental factors.1, 2, 3, 4, 5 In addition, adult height is easily and accurately measured, and remains relatively stable over much of an individual's lifespan.6 Furthermore, height is the result of many growth and development processes and is associated with various human diseases. Thus, genetic variants affecting height variation might provide new insight into human growth and development, as well as the genetic architecture of several human diseases.7
Many genetic linkage studies have attempted to identify the variants influencing the normal variations in human height. Quantitative trait loci for adult height were scattered across the genome with minimal overlap.8 Furthermore, the resolution of these studies was low, and they have not identified genetic variants that explain the linkage signals.9 With the development of genome-wide association (GWA) studies, progress in the field of human genetics has been rapid. In 2008, three consortia of research groups reported 54 genetic variants associated with adult height using data from GWA studies.10, 11, 12 However, these studies were performed in Caucasian populations whose genetic background differs from that of Asians. Therefore, it is unclear whether the associated loci have similar roles in Asian populations, particularly because a recent GWA study performed in a Chinese population revealed markedly different results from those obtained in a Caucasian population.13
Recently, we performed a GWA study of eight quantitative traits (such as, human height) of biomedical importance in the Korean population. The study identified eight loci that were significantly associated with height, including HMGA1, SPAG17, ZBTB38, PLAG1, FBP2, TBX2, EFEMP1 and LTBP1.14 In our current study, we analyzed the same data using different analysis approaches to identify additional genetic variants influencing adult height in Koreans. Here, we report newly identified associations between single-nucleotide polymorphisms (SNPs) and height variation. In addition, we investigated the contribution of height loci to idiopathic short stature (ISS), a condition in which the height of the individual is more than two standard deviations (s.d.) less than the corresponding mean height for a given age, sex and population (that is, equivalent to 2.3% of the shortest individuals of the population) for unknown or hereditary reasons.15
Materials and methods
Subjects
Participants in the GWA study were recruited from two community-based cohorts (that is, the Ansung and Ansan cohorts) in the Gyeonggi Province of South Korea. Both cohorts were involved in the Korean Genome Epidemiology Study (KoGES), a longitudinal prospective study initiated in 2001. The Ansung and Ansan cohorts consisted of 5018 and 5020 participants, respectively, ranging in age from 40 to 69 years. These cohorts adhered to the identical investigational strategy during every 2-year examination. More than 260 traits have been extensively examined through epidemiological surveys, physical examinations and laboratory tests.14
The ISS patients were recruited from the Department of Pediatrics at the Asan Medical Center in Seoul, Korea. The study group included 128 children with ISS, all of whom met the following criteria: (a) height less than −2 standard deviation score (SDS) relative to age- and sex-matched controls, (b) birth weight more than −2 SDS relative to gestational age- and sex-matched controls, (c) normal height velocity (HV) for 1 year or normal growth hormone (GH) response on two provocative tests in patients with slow HV and (d) no earlier treatment with GH or other anabolic agents. Children with other causes of short stature (for example, genetic, syndromic or organic conditions) were excluded from the study. Control samples used in the case–control association study were obtained from the adult health cohort of the general population in Korea. These controls had no history of disease and were provided by the Biobank for Health Sciences at the Center for Genome Sciences in Seoul, Korea. Initially, our control population consisted of 828 individuals. However, 41 individuals were excluded from the association analysis because they ranked in the bottom 5% of the height distribution of 828 people. Basic anthropometric data of the study subjects are presented in Supplementary Table 1. These studies were approved by the Institutional Review Board of the involved institutions, and written informed consent was obtained from all subjects and from the parents of all ISS patients.
Genotyping and quality control
Genotyping methods and quality control steps for the GWA study were performed as previously described.14 Briefly, 10 004 genomic DNA samples were genotyped using the Affymetrix Genome-Wide Human SNP array 5.0 (Affymetrix, Santa Clara, CA, USA). Bayesian Robust Linear Modeling using the Mahalanobis Distance (BRLMM) Genotyping Algorithm (Affymetrix) was used for the genotype calling of 500 568 SNPs. Samples with high missing genotype call rates (>4%, n=401), high heterozygosity (>30%, n=11), inconsistency in sex (n=41), high identity-by-state values (>0.80, n=601) and any kinds of cancer (n=101) were excluded from the analysis. A total of 8842 individuals were included in subsequent analyses. To filter the SNP markers, we also excluded SNP markers with missing call rates ⩾1% (n=104 831) and minor allele frequencies (MAF) <0.001 (n=63 380). After filtering, 334 546 SNPs were included in the analysis.
The Illumina VeraCode GoldenGate Assay kit (Illumina, San Diego, CA, USA) was used to perform the genotyping of SNPs selected for association analysis with ISS, according to the manufacturer's instructions. Genotype clustering and calling were performed using BeadStudio software (Illumina). The overall genotype success rate was 99.88% and one sample with a high missing call rate (9.9%) was excluded from subsequent analyses.
Statistical analysis
Statistical analyses were performed using the PLINK (version 1.05) (http://pngu.mgh.harvard.edu/~purcell/plink/)16 and SPSS programs (version 12.0) (SPSS, Chicago, IL, USA). The GWA study samples exhibited normal height distributions, and individual raw values were used in the association analyses. Height associations were assessed using linear regression analysis adjusted for sex and age under additive, dominant and recessive models for all individuals (n=8842). To test the association with ISS, the χ2-test and logistic regression analysis were used to compare allele and genotype frequencies and to estimate the odds ratios in the case and control subjects.
Results
Identification of loci associated with adult height
We recently described eight loci (HMGA1, SPAG17, ZBTB38, PLAG1, FBP2, TBX2, EFEMP1 and LTBP1) that are significantly associated with adult height in the Korean population (n=8842).14 Those results were analyzed by linear regression analysis using an additive model. In this study, we analyzed the same data using a different approach by using three different genetic models to search for additional genetic loci affecting the adult height of Koreans. To determine common alleles associated with height, linear regression analysis adjusted for sex and age was performed under additive, dominant and recessive models in all individuals (n=8842), as previously described.14 The results of our linear regression analysis are shown in Table 1. A total of 36 SNPs in 21 loci had significant effects (P<1 × 10−5). Among those 36 SNPs, 28 SNPs in 15 loci (LTBP1, EFEMP1, ZBTB38, HMGA1, SUPT3H, PLAG1, EXT1, FREM1, FBP2, PALM2-AKAP2, NUP37-PMCH, IGF1, KRT20, TBX4 and ANKRD60) were commonly significant with P<0.01 in both Ansung and Ansan areas (Supplementary Table 2). Among the 15 loci associated with height, seven (LTBP1, EFEMP1, ZBTB38, HMGA1, PLAG1, FBP2 and TBX4) had been previously described in Caucasians and/or our previous report14 and eight were newly identified for the first time in Koreans.
We then compared the height-associated SNPs in the Korean population with those previously reported in GWA studies of Caucasian individuals (Supplementary Table 3). Eighteen loci (SPAG17, ZNF678, EFEMP1, ZBTB38, HHIP, LCORL, BAT3, HMGA1, PLAG1, CHCHD7, PTCH1, HMGA2, SOCS2, ACAN, TBX4, CABLES1, BMP2 and GDF5-UQCC) were consistent with earlier studies (P<0.05) and 15 previously reported loci (SCMH1, IHH, ANAPC13, C6orf106, LOC387103, GNA12, CDK6, PXMP3, ZNF462, DLEU7, TRIP11, ADMTSL3, ADAMTS17, NOG and DYM) did not show significant association with human height in the Korean population. The remaining loci could not be validated because they were not included in the Affymetrix Genome-Wide Human SNP Array 5.0. The MAF in Caucasian and Korean population were not significantly different with regard to replicated and non-replicated SNPs (data not shown).
Combined effects of height-associated SNPs
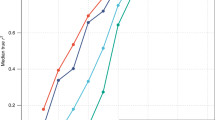
To assess the combined effects of the 15 significant SNPs on adult height (Table 1), we counted the number of height-increasing alleles by applying the genetic mode of the significant SNPs for each individual with the complete genotypes for these SNPs. We then classified the individuals according to the number of height-increasing alleles, and determined the average height for each group. Figure 1 shows the linear increase in the average height with increasing numbers of ‘tall’ alleles. Altogether, the 15 SNPs accounted for approximately 1.0% of the height variation. Males and females with at least 8 ‘tall’ alleles (5.1%) were 3.0 and 4.4 cm shorter, respectively, than those with at least 19 ‘tall’ alleles (4.2%).

Combined effects of 15 significant single-nucleotide polymorphisms (SNPs) on adult height. The number of height-increasing alleles was counted in our samples (n=8842), along with the complete genotype for the 15 significant SNPs, and individuals were classified according to the number of ‘tall’ alleles. For each group, the mean±95% confidence interval (95% CI) was plotted separately for males and females. The black regression line indicates the mean height of each group (that is, including all individuals), and shows that each additional ‘tall’ allele increases height by 0.3 cm. The bars represent the proportion of the sample in each group.
Identification of loci associated with ISS
To search for loci associated with ISS, we selected a total of 44 loci that included newly identified SNPs in Koreans and previously reported SNPs associated with human height. The SNP sites were genotyped in 128 ISS patients and 787 normal controls whose height was greater than that of the bottom 5% of 828 normal individuals. Genetic association analysis identified five genes (SPAG17, KBTBD8, HHIP, HIST1H1D and ACAN) that were significantly associated with ISS (uncorrected P<0.05) (Table 2), indicating that some height-associated genes are involved in conditions such as ISS that are characterized by extreme human height.
Discussion
We performed a GWA analysis of adult height in 8842 Korean individuals and identified 15 genetic regions with significant associations at the P-value <1 × 10−5 threshold in all 8842 individuals and subgroup analysis with P-value <0.01 in both the Ansung and Ansan cohorts. We chose this arbitrary threshold as suggestive association due to the relatively small sample size in our study, even though this threshold does not reach genome-wide significance P-value of 1.5 × 10−7 (in case of P-value <0.05 with 334 546 SNPs used in analysis after SNP filtering). In linear regression analysis, we controlled for sex and age as covariates. In addition, we subdivided our sample into two areas, performed the same analysis, and selected significant SNPs (P<0.01) that were common to both areas. Among the 15 suggestive loci, seven (LTBP1, EFEMP1, ZBTB38, HMGA1, PLAG1, FBP2 and TBX4) had previously been described in Caucasians and/or our previous report,14 whereas eight were described for the first time in Koreans. Our loose significance threshold had limited power to confirm the associations for some loci. However, many of the newly discovered loci have clear biological functions related to height.
Among the eight loci (SUPT3H, EXT1, FREM1, PALM2-AKAP2, NUP37-PMCH, IGF1, KRT20 and ANKRD60) that were newly identified in the Korean population, the EXT1 gene encodes an endoplasmic reticulum-resident type II transmembrane glycosyltransferase involved in the chain elongation step of heparan sulfate biosynthesis.17 Mutations in this gene cause chondrosarcoma18 and the type I form of multiple exostoses, an autosomal dominant disorder characterized by multiple projections of bone capped by cartilage. This condition can lead to skeletal abnormalities, short stature and malignant transformation of exostoses to chondrosarcomas or osteosarcomas.19 The PALM2-AKAP2 mRNA is a naturally occurring co-transcribed product of the neighboring PALM2 and AKAP2 genes. The significance of this co-transcribed mRNA and the function of its protein product have not yet been determined. However, a balanced translocation t(7;9)(p14.1;q31.3) that completely disrupted the AKAP2 gene caused a complex phenotype comprising Kallmann's syndrome and bone anomalies.20 IGF1 belongs to a family of proteins that mediate growth and development. Specifically, IGF1 mediates several growth-promoting effects of GHs. Defects in this gene result in a deficiency in insulin-like growth factor-1.21 In addition, although the sample size is limited to detect the same level of significance in 828 sample set, we confirmed that PALM2-AKAP2 (rs7032940), IGF1 (rs1520223) and EFEMP1 (rs3791675) loci were significantly associated with human height (P<0.05) (Supplementary Table 4).
Comparison of our results with previous findings in Caucasian populations revealed that many loci associated with adult height overlap: 18 of the 34 loci tested were replicated (P<0.05) (Supplementary Table 3). This conflicts with recently published results in a Chinese GWA study.13 The authors of the Chinese study reported that 6 of the 44 SNPs identified in Caucasian individuals were also significant in a Chinese population (P<0.1, with the same direction of effect).13 However, the number of subjects included in the Chinese GWA study was limited (n=618), so the studies cannot be directly compared. However, we observed ethnic differences in MAFs of the SNPs present in both Caucasians and Koreans. Therefore, it is likely that there are ethnic differences in genetic effects on human height.
In this study, we assessed the contribution of height-associated loci to ISS. We tested the association of 44 loci in 128 patients with ISS and 787 normal control individuals. Five loci (SPAG17, KBTBD8, HHIP, HIST1H1D and ACAN) were associated with ISS (uncorrected P<0.05). The HHIP gene encodes a regulatory component in the Hedgehog signaling pathway. Ectopic expression of Hip in transgenic mice results in severe skeletal defects similar to those observed in Indian Hedgehog (IHH) mutants.22 The HIST1H1D gene encodes a member of the H1 class of histones.23 It is currently unclear how genetic variants at this locus modulate height. However, several SNPs associated with height in Caucasian populations are involved in chromatin structure and regulation.7 The ACAN gene is a member of the aggrecan/versican proteoglycan family. The encoded protein is an integral part of the extracellular matrix in cartilaginous tissue. Mutations in this gene cause an autosomal dominant type of spondyloepiphyseal dysplasia, designated SED type Kimberley (SEDK). This disorder is characterized by proportionate short stature (below the fifth percentile for age), with a stocky habitus and progressive osteoarthropathy of the weight-bearing joints.24
In summary, we examined genome-wide scan data from 8842 individuals and identified 15 loci that influence adult height. In addition, we also identified five genetic loci associated with extreme short stature ISS. This study provides insight into new candidate regions involved in human growth and development. Further replication of our results in other cohorts, as well as fine mapping by resequencing and functional studies, will help identify the true causative variants and will provide a better understanding of the mechanisms of human growth and development.
References
Carmichael, C. M. & McGue, M. A cross-sectional examination of height, weight, and body mass index in adult twins. J. Gerontol. A. Biol. Sci. Med. Sci. 50, B237–B244 (1995).
Hirschhorn, J. N., Lindgren, C. M., Daly, M. J., Kirby, A., Schaffner, S. F., Burtt, N. P. et al. Genomewide linkage analysis of stature in multiple populations reveals several regions with evidence of linkage to adult height. Am. J. Hum. Genet. 69, 106–116 (2001).
Phillips, K. & Matheny, Jr A. P. Quantitative genetic analysis of longitudinal trends in height: preliminary results from the Louisville Twin Study. Acta. Genet. Med. Gemellol. (Roma) 39, 143–163 (1990).
Preece, M. A. The genetic contribution to stature. Horm. Res. 45 (Suppl 2), 56–58 (1996).
Silventoinen, K., Kaprio, J., Lahelma, E. & Koskenvuo, M. Relative effect of genetic and environmental factors on body height: differences across birth cohorts among Finnish men and women. Am. J. Public Health 90, 627–630 (2000).
Mathias, R. A., Roy-Gagnon, M. H., Justice, C. M., Papanicolaou, G. J., Fan, Y. T., Pugh, E. W. et al. Comparison of year-of-exam- and age-matched estimates of heritability in the Framingham Heart Study data. BMC Genet 4 (Suppl 1), S36 (2003).
Weedon, M. N. & Frayling, T. M. Reaching new heights: insights into the genetics of human stature. Trends Genet. 24, 595–603 (2008).
Perola, M., Sammalisto, S., Hiekkalinna, T., Martin, N. G., Visscher, P. M., Montgomery, G. W. et al. Combined genome scans for body stature in 6602 European twins: evidence for common Caucasian loci. PLoS Genet. 3, e97 (2007).
Visscher, P. M. Sizing up human height variation. Nat. Genet. 40, 489–490.
Gudbjartsson, D. F., Walters, G. B., Thorleifsson, G., Stefansson, H., Halldorsson, B. V., Zusmanovich, P. et al. Many sequence variants affecting diversity of adult human height. Nat. Genet. 40, 609–615 (2008).
Lettre, G., Jackson, A. U., Gieger, C., Schumacher, F. R., Berndt, S. I., Sanna, S. et al. Identification of ten loci associated with height highlights new biological pathways in human growth. Nat. Genet. 40, 584–591 (2008).
Weedon, M. N., Lango, H., Lindgren, C. M., Wallace, C., Evans, D. M., Mangino, M. et al. Genome-wide association analysis identifies 20 loci that influence adult height. Nat. Genet. 40, 575–583 (2008).
Lei, S. F., Yang, T. L., Tan, L. J., Chen, X. D., Guo, Y., Guo, Y. F. et al. Genome-wide association scan for stature in Chinese: evidence for ethnic specific loci. Hum. Genet. 125, 1–9 (2009).
Cho, Y. S., Go, M. J., Kim, Y. J., Heo, J. Y., Oh, J. H., Ban, H. J. et al. A large-scale genome-wide association study of Asian populations uncovers genetic factors influencing eight quantitative traits. Nat. Genet. 41, 527–534 (2009).
Wit, J. M., Clayton, P. E., Rogol, A. D., Savage, M. O., Saenger, P. H. & Cohen, P. Idiopathic short stature: definition, epidemiology, and diagnostic evaluation. Growth Horm. IGF Res. 18, 89–110 (2008).
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
McCormick, C., Leduc, Y., Martindale, D., Mattison, K., Esford, L. E., Dyer, A. P. et al. The putative tumour suppressor EXT1 alters the expression of cell-surface heparan sulfate. Nat. Genet. 19, 158–161 (1998).
Hecht, J. T., Hogue, D., Wang, Y., Blanton, S. H., Wagner, M., Strong, L. C. et al. Hereditary multiple exostoses (EXT): mutational studies of familial EXT1 cases and EXT-associated malignancies. Am. J. Hum. Genet. 60, 80–86 (1997).
Peterson, H. A. Multiple hereditary osteochondromata. Clin. Orthop. Relat. Res. 239, 222–230 (1989).
Panza, E., Gimelli, G., Passalacqua, M., Cohen, A., Gimelli, S., Giglio, S. et al. The breakpoint identified in a balanced de novo translocation t(7;9)(p14.1;q31.3) disrupts the A-kinase (PRKA) anchor protein 2 gene (AKAP2) on chromosome 9 in a patient with Kallmann syndrome and bone anomalies. Int. J. Mol. Med. 19, 429–435 (2007).
Walenkamp, M. J., Karperien, M., Pereira, A. M., Hilhorst-Hofstee, Y., van Doorn, J., Chen, J. W. et al. Homozygous and heterozygous expression of a novel insulin-like growth factor-I mutation. J. Clin. Endocrinol. Metab. 90, 2855–2864 (2005).
Chuang, P. T. & McMahon, A. P. Vertebrate Hedgehog signalling modulated by induction of a Hedgehog-binding protein. Nature 397, 617–621 (1999).
Albig, W., Drabent, B., Kunz, J., Kalff-Suske, M., Grzeschik, K. H. & Doenecke, D. All known human H1 histone genes except the H1(0) gene are clustered on chromosome 6. Genomics 16, 649–654 (1993).
Anderson, I. J., Tsipouras, P., Scher, C., Ramesar, R. S., Martell, R. W. & Beighton, P. Spondyloepiphyseal dysplasia, mild autosomal dominant type is not due to primary defects of type II collagen. Am. J. Med. Genet. 37, 272–276 (1990).
Acknowledgements
This work was supported by the Korea Science and Engineering Foundation (KOSEF) grant funded by the Korea government (MEST) (No. 2009-0075450), a grant from the Ministry of Health & Welfare of the Republic of Korea (01-PJ10-PG6-01GN15-001) and a grant from the Korea Centers for Disease Control and Prevention (No. 2008-S6-E-001). The work of JK Lee was partially supported by a grant (No. 2009-419) from the Asan Institute for Life Sciences in Seoul, Korea.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website (http://www.nature.com/jhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Kim, JJ., Lee, HI., Park, T. et al. Identification of 15 loci influencing height in a Korean population. J Hum Genet 55, 27–31 (2010). https://doi.org/10.1038/jhg.2009.116
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2009.116
Keywords
This article is cited by
-
Whole genome sequencing of mouse lines divergently selected for fatness (FLI) and leanness (FHI) revealed several genetic variants as candidates for novel obesity genes
Genes & Genomics (2024)
-
A QTL for conformation of back and croup influences lateral gait quality in Icelandic horses
BMC Genomics (2021)
-
Complex Phenotypes: Mechanisms Underlying Variation in Human Stature
Current Osteoporosis Reports (2019)
-
Association of human height-related genetic variants with familial short stature in Han Chinese in Taiwan
Scientific Reports (2017)
-
Multi-strategy genome-wide association studies identify the DCAF16-NCAPG region as a susceptibility locus for average daily gain in cattle
Scientific Reports (2016)
