
Abstract
Mice with a deletion of Tgf-β3 (−/−) and association studies in humans of different ethnicities support the involvement of TGFB3 in the etiology of orofacial clefts. In this study, we investigated the relevance of TGFB3 in the development of cleft lip and palate (CL/P) among 204 triads of central European origin. Transmission-disequilibrium test (TDT) analysis revealed no significant transmission distortions for each marker alone, and none for any possible haplotypes. However, we found strong evidence for parent-of-origin effects, with lower risk of maternal transmission compared with paternal transmission [I M = 0.38; confidence interval (CI): 0.17–0.86] of the risk allele T to an affected offspring at marker rs2300607. This is also expressed in an increased risk of heterozygous children having the T allele inherited from the father (R P = 3.47; CI: 1.32–9.11). Our data support the involvement of TGFB3 in the development of oral clefts in patients of central European origin.
Similar content being viewed by others

Introduction
The genetic background of nonsyndromic orofacial clefts is complex, and most causal genes remain to be identified. The majority of pertinent candidate genes have been deducted from murine knockout models. One of these genes is the transforming growth factor-beta 3 gene TGFB3 (NM003239.1 NP003230.1), located on human chromosome 14q24. TGFB3 is expressed in medial-edge epithelium cells of palatal shelves (Gato et al. 2002). Mice with a genetic deletion of Tgf-β3 (−/−) present with either complete cleft palate or severe partial cleft palate (Kaartinen et al. 1995; Proetzel et al. 1995). Genetic association studies in humans of different ethnicities suggest TGFB3 may be involved in the formation of orofacial clefts (Maestri et al. 1997; Lidral et al. 1998; Romitti et al. 1999; Mitchell et al. 2001; Sato et al. 2001; Beaty et al. 2002; Scapoli et al. 2002; Jugessur et al. 2003; Kim et al. 2003; Slayton et al. 2003; Vieira et al. 2003; Suzuki et al. 2004), although negative studies have also been reported (Lidral et al. 1997; Tanabe et al. 2000; Beaty et al. 2001; Morkūniené et al. 2007). Until recently, most studies used a CA repeat located 61.215 bp upstream from translation starting point of TGFB3 as well as a VNTR marker in the 5′ untranslated region (UTR) (Δ at −21083 to −21086 (AGAGGG repeat)) and with X5.1 a T > C substitution [ref. single nucleotide polymorphism (SNP) rs3917200] at position −24 relative to intron 4/exon 5 junction to test for involvement of the gene (Lidral et al. 1998; Beaty et al. 2002). Recently, the study by Ichikawa et al. (2006) aimed at a comprehensive coverage of the gene and tested a total of 23 SNP in a Japanese cleft lip and palate (CL/P) population. These authors observed significant association, with P < 0.01 at four SNPs and P < 0.05 at an additional five SNPs. As this is the most convincing human study of TGFB3 in CL/P published to date, we investigated the relevance of their findings for CL/P patients of a different ethnic background, namely, patients of central European origin. In addition, we searched for parent-of-origin effects, which have been previously described for candidate genes associated with nonsyndromic oral clefts (Scapoli et al. 2002; Rubini et al. 2005).
Materials and methods
Sample
In our study, we included 204 patients with nonsyndromic CL/P and their parents. Triads were recruited throughout Germany from hospitals and non-hospital-based institutions. Ethical approval was obtained from ethics committees of the relevant medical facilities. All individuals and, in cases of children younger than 18 years, parents gave written informed consent for participation. To rule out syndromic CL/Ps, each patient or the parents were asked to complete a detailed questionnaire on psychomotor development and general health status of the affected person. Furthermore, a thorough clinical examination was performed in each patient and his/her parents by one of two clinical geneticists (see also Birnbaum et al. 2007). The ethnic background of the patients was assessed by identifying the origin of their grandparents. Only patients whose four grandparents were born in central Europe were classified as being of central European origin. Peripheral venous blood was taken for DNA extraction from all participants. The patient sample comprised 121 male subjects and 83 female subjects. In 176 families, both parents participated in the study (complete triads); in 26 family units, only the mother was available, and in two only the father (28 incomplete triads) was available. Of the 204 triads, 14 mothers and 21 fathers had a nonsyndromic CL/P themselves.
Genotype analysis
To test for association, we used three SNPs (IVS1 +2118, IVS1 +5321, and IVS1 −1572), which were significantly associated with CL/P in the study by Ichikawa et al. (2006) and which had minor allele frequencies >0.05 in Japanese and central Europeans (see NCBI: http://www.ncbi.nlm.nih.gov/SNP). In Ichikawa et al.’s study (2006), markers IVS1 +2118 and IVS1 +5321 displayed the most significant association in their single-marker analysis. In the two marker haplotype analysis, the combination of IVS1 +5321 and IVS1 −1572 showed the most significant result. Genotyping of SNP rs2268626 (IVS1 +2118) and rs2268625 (IVS1 −1572) was accomplished using TaqMan assay on an ABI 7500 (Applied Biosystems) according to predesigned conditions. Because SNP rs2300607 (IVS1 +5321) was located in a highly polymorphic region, allele screening was performed with a polymerase chain reaction (PCR)-based restriction assay. PCR was performed using the FastStart Taq DNA Polymerase kit (Roche, Applied Science, Indianapolis, IN, USA) following standard conditions with the use of modified primers (4F: 5′-TCAGCCTGGACAACATAGGGAGGACC-3′; 4R: 5′-TATGTACATTTTCTTTGATCTCCCAGG-3′). If the minor allele T was present in the amplified product (405 bp), two ApoI (Fermentas ER1382) restriction sites resulted in fragments of 51, 140, and 214 bp in size. The digested PCR products were separated on 3% agarose gels.
Statistical analysis
Genotype distribution obtained by combining the two nontransmitted parental alleles in each family was tested for deviations from Hardy–Weinberg equilibrium by Weir’s exact test (Weir 1996). We used the transmission disequilibrium test (TDT) to check for distortion of the inheritance of alleles from parents to affected offspring (Spielman et al. 1993). Haplotype analysis was performed with the software FAMHAP (Becker und Knapp 2004). A log-linear model (Weinberg et al. 1998; Weinberg 1999) allows screening for genotype effects of child and mother as well as parent-of-origin effects. The program LEM (http://www.uvt.nl/faculteiten/fsw/organisatie/departementen/mto/software2.html) (Vermunt 1997; van den Oord and Vermunt 2000) was applied to estimate the relative risks (RR) of Weinberg’s model and to obtain corresponding 95% confidence intervals (CI). CI not covering the value of 1 provide evidence of a significant effect at α = 5%.
Results
For all SNPs, the distribution of genotypes obtained by combining the two nontransmitted parental alleles was consistent with Hardy–Weinberg equilibrium (data not shown). TDT analysis revealed no significant transmission distortions for each marker alone and none for any of the possible haplotypes (Tables 1 and 2). Our sample had power of approximately 80% to detect (at α = 5%) a transmission distortion associated with a RR Ψ2 = 1.8 for the homozygous genotype and Ψ1 = 1.0 for the heterozygous genotype (recessive mode of inheritance) and for RR of Ψ2 = 2.56 and Ψ1 = 1.6 (multiplicative model).
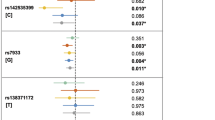
Analysis according to Weinberg’s log-linear model (Weinberg 1999) revealed that a paternally derived copy of allele T at marker rs2300607 (IVS1 +5321) induced a greater increase in risk than did a maternally derived copy. Table 3 presents estimates of RR and corresponding 95% CI for these parameters. Here, parameters S 1 or S 2, respectively, means RR of one copy or two copies, respectively, against no copy of the variant allele in the mother. Two copies of a variant in a child increased the RR by a factor of R 2. When considering parent-of-origin effects, R P is the relative risk associated with a single copy inherited from the father, and R P × I M represents the risk associated with a single copy inherited from the mother, i.e., I M = 0.38 < 1 (CI: 0.17–0.86, P = 0.019), which reflects a decreased probability of a maternally derived copy compared with a paternally derived copy. In addition, our analysis demonstrated an elevated R P = 3.47 (CI: 1.32–9.11), i.e., an increased risk for children with only one paternally inherited allele.
Discussion
The process of palatal fusion is controlled by interactive signaling from the mesenchyme to the epithelium, which is mediated by growth factors and extracellular matrix (ECM) proteins (Baroni et al. 2003). One of the key components in palatal fusion is transforming growth factor β3 (Tgf-β3) (Nogai et al. 2008). Tgf-β3 expression, which is coordinated temporally during palatogenesis (Britto et al. 2002), determines ECM macromolecule overaccumulation by stimulating ECM neosynthesis and by inhibiting many enzymes implicated in ECM degradation (Blavier et al. 2001). Consequently, reduced activity of Tgf-β3 may increase the risk for oral clefting. Consistent with this hypothesis, Tgf-β3 −/− knockout mice present with a developmental defect of the secondary palate (Proetzel et al. 1995). In humans, however, evidence for any involvement of the TGFB3 gene in development of oral clefts has remained inconclusive, with reports of significant (Maestri et al. 1997; Lidral et al. 1998; Romitti et al. 1999; Mitchell et al. 2001; Sato et al. 2001; Beaty et al. 2002; Scapoli et al. 2002; Jugessur et al. 2003; Kim et al. 2003; Slayton et al. 2003; Vieira et al. 2003; Suzuki et al. 2004) as well as negative (Lidral et al. 1997; Tanabe et al. 2000; Beaty et al. 2001; Morkūniené et al. 2007) associations among different populations with nonsyndromic orofacial clefts. In this study, we investigated whether the results of Ichikawa et al. (2006), who conducted the most comprehensive study of TGFB3 to date, were also apparent in CL/P families of central European descent. We used a case–parent triad design to avoid undetected ethnic stratification as a cause of false positive results.
In our sample of 204 families, we observed no significant transmission distortion, neither at the level of individual markers nor at the level of haplotypes (Tables 1 and 2).
Furthermore, because parent-of-origin effects have been previously described for oral-cleft candidate genes (Scapoli et al. 2002; Rubini et al. 2005), we tested TGFB3 variants for evidence of genomic imprinting. Interestingly, however, we found evidence of a lower risk of maternal transmission compared with paternal transmission (I M = 0.38; CI: 0.17–0.86) of allele T to the affected offspring at marker rs2300607 (IVS1 +5321) (Table 3). This is also expressed as an increased risk of heterozygous children having the T allele inherited from the father (R P = 3.47; CI: 1.32–9.11 and R 2 = 1.98; CI: 0.76–5.21). According to our data, epigenetic modulation of the paternal inherited allele could lead to disturbance of palatal fusion during embryogenesis resulting in greater susceptibility to CL/P in the offspring.
Genomic imprinting modulates gene expression by turning specific alleles of a gene on or off depending on which parent transmits them. Both, genetic and environmental factors can affect the imprinting process and alter the level of expression of imprinted genes (Jirtle et al. 2000).
In general, epigenetic effects are increasingly recognized as an important source of variation in complex traits. Principle among these effects is genomic imprinting, which has generally been examined in analyses of complex traits by testing for parent-of-origin-dependent effects of alleles. However, we acknowledge that in most of these analyses, maternal effects are confounded with genomic imprinting because they can produce the same patterns of phenotypic variation expected for various forms of imprinting (Hager et al. 2008).
In summary, our analysis suggests a role for TGFB3 in the etiology of CL/P among the central European population if the risk allele is inherited from the father. Although these data suggest an intriguing role of TGFB3 in the development of oral clefts, the hypothesis still requires a convincing degree of support from independent studies. Also, functional studies are warranted to show any effect of allelic variants on activation of Tgf-β3 and to provide experimental evidence for TGFB3 being subjected to genomic imprinting.
References
Baroni T, Carinci P, Bellucci C, Lilli C, Becchetti E, Carinci F, Stabellini G, Pezzetti F, Caramelli E, Tognon M, Bodo M (2003) Cross-talk between interleukin-6 and transforming growth factor-beta3 regulates extracellular matrix production by human fibroblasts from subjects with non-syndromic cleft lip and palate. J Periodontol 74:1447–1453
Beaty TH, Wang H, Hetmanski JB, Fan YT, Zeiger JS, Liang KY, Chiu YF, Vanderkolk CA, Seifert KC, Wulfsberg EA, Raymond G, Panny SR, McIntosh I (2001) A case-control study of nonsyndromic oral clefts in Maryland. Ann Epidemiol 11:434–442
Beaty TH, Hetmanski JB, Zeiger JS, Fan YT, Liang KY, VanderKolk CA, McIntosh I (2002) Testing candidate genes for non-syndromic oral clefts using a case-parent trio design. Genet Epidemiol 22:1–11
Becker T, Knapp M (2004) A powerful strategy to account for multiple testing in the context of haplotype analysis. Am J Hum Genet 75:561–570
Birnbaum S, Reutter H, Mende M, Díaz-Lacava A, Henschke H, Bergé SJ, Braumann B, Lauster C, Hemprich A, Wenghoefer M, Saffar M, Reich RH, Scheer M, Kramer F-J, Knapp M, Mangold E (2007) A family-based association study in central Europeans: no evidence for the cystathionine beta-synthase c.844ins68 gene variant as a risk factor for non-syndromic cleft lip and palate. Am J Med Genet A 143:205–207
Britto JA, Evans RD, Hayward RD, Jones BM (2002) Toward pathogenesis of Apert cleft palate: FGF, FGFR, and TGF beta genes are differentially expressed in sequential stages of human palatal shelf fusion. Cleft Palate Craniofac J 39:332–340
Blavier L, Lazaryev A, Groffen J, Heisterkamp N, DeClerck YA, Kaartinen V (2001) TGF-beta3-induced palatogenesis requires matrix metalloproteinases. Mol Biol Cell 12:1457–1466
Gato A, Martinez ML, Tudela C, Alonso I, Moro JA, Formoso MA, Ferguson MW, Martinez-Alvarez C (2002) TGF-beta(3)-induced chondroitin sulphate proteoglycan mediates palatal shelf adhesion. Dev Biol 250:393–405
Hager R, Cheverud JM, Wolf JB (2008) Maternal effects as the cause of parent-of-origin effects that mimic genomic imprinting. Genetics 178:1755–1762
Ichikawa E, Watanabe A, Nakano Y, Akita S, Hirano A, Kinoshita A, Kondo S, Kishino T, Uchiyama T, Niikawa N, Yoshiura K (2006) PAX9 and TGFB3 are linked to susceptibility to nonsyndromic cleft lip with or without cleft palate in the Japanese: population-based and family-based candidate gene analyses. J Hum Genet 51:38–46
Jirtle RL, Sander M, Barrett JC (2000) Genomic imprinting and environmental disease susceptibility. Environ Health Perspect 108:271–278
Jugessur A, Lie RT, Wilcox AJ, Murray JC, Taylor JA, Saugstad OD, Vindenes HA, Abyholm F (2003) Variants of developmental genes (TGFA, TGFB3, and MSX1) and their associations with orofacial clefts: a case-parent triad analysis. Genet Epidemiol 24:230–239
Kaartinen V, Voncken JW, Shuler C, Wartburton D, Bu D, Heisterkamp N, Groffen J (1995) Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat Genet 11:415–421
Kim MH, Kim HJ, Choi JY, Nahm DS (2003) Transforming growth factor-beta3 gene SfaN1 polymorphism in Korean nonsyndromic cleft lip and palate patients. J Biochem Mol Biol 36:533–537
Lidral AC, Murray JC, Buetow KH, Basart AM, Schearer H, Shiang R, Naval A, Layda E, Magee K, Magee W (1997) Studies of the candidate genes TGFB2, MSX1, TGFA, and TGFB3 in the etiology of cleft lip and palate in the Philippines. Cleft Palate Craniofac J 34:1–6
Lidral AC, Romitti PA, Basart AM, Doetschman T, Leysens NJ, Daack-Hirsch S, Semina EV, Johnson LR, Machida J, Burds A, Parnell TJ, Rubenstein JL, Murray JC (1998) Association of MSX1 and TGFB3 with nonsyndromic clefting in humans. Am J Hum Genet 63:557–568
Maestri NE, Beaty TH, Hetmanski J, Smith EA, McIntosh I, Wyszynski DF, Liang KY, Duffy DL, Vanderkolk C (1997) Application of transmission disequilibrium tests to nonsyndromic oral clefts: including candidate genes and environmental exposures in the models. Am J Med Genet 73:337–344
Mitchell LE, Murray JC, O’Brien S, Christensen K (2001) Evaluation of two putative susceptibility loci for oral clefts in the Danish population. Am J Epidemiol 153:1007–1015
Morkūniené A, Steponaviciūt D, Utkus A, Kucinskas V (2007) Few associations of candidate genes with nonsyndromic orofacial clefts in the population of Lithuania. J Appl Genet 48:89–91
Nogai H, Rosowski M, Grün J, Rietz A, Debus N, Schmidt G, Lauster C, Janitz M, Vortkamp A, Lauster R (2008) Follistatin antagonizes transforming growth factor-beta3-induced epithelial-mesenchymal transition in vitro: implications for murine palatal development supported by microarray analysis. Differentiation 76(4):404–416
Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, Ding J, Ferguson MW, Doetschman T (1995) Transforming growth factor-beta 3 is required for secondary palate fusion. Nat Genet 11:409–414
Romitti PA, Lidral AC, Munger RG, Daack-Hirsch S, Burns TL, Murray JC (1999) Candidate genes for nonsyndromic cleft lip and palate and maternal cigarette smoking and alcohol consumption: evaluation of genotype-environment interactions from a population-based case-control study of orofacial clefts. Teratology 59:39–50
Rubini M, Brusati R, Garattini G, Magnani C, Liviero F, Bianchi F, Tarantino E, Massei A, Pollastri S, Carturan S, Amadori A, Bertagnin E, Cavallaro A, Fabiano A, Franchella A, Calzolari E (2005) Cystathionine beta-synthase c.844ins68 gene variant and non-syndromic cleft lip and palate. Am J Med Genet A 136:368–372
Sato F, Natsume N, Machido J, Suzuki S, Kawai T (2001) Association between transforming growth factor beta 3 and cleft lip and/or palate in the Japanese population. Plast Reconstr Surg 107:1909–1910
Scapoli L, Martinelli M, Pezzetti F, Carinci F, Bodo M, Tognon M, Carinci P (2002) Linkage disequilibrium between GABRB3 gene and nonsyndromic familial cleft lip with or without cleft palate. Hum Genet 110:15–20
Slayton RL, Williams L, Murray JC, Wheeler JJ, Lidral AC, Nishimura CJ (2003) Genetic association studies of cleft lip and/or palate with hypodontia outside the cleft region. Cleft Palate Craniofac J 40:274–279
Spielman RS, McGinnis RE, Ewens WJ (1993) Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet 52:506–516
Suzuki Y, Jezewski PA, Machida J, Watanabe Y, Shi M, Cooper ME, Viet le T, Nguyen TD, Hai H, Natsume N, Shimozato K, Marazita ML, Murray JC (2004) In a Vietnamese population, MSX1 variants contribute to cleft lip and palate. Genet Med 6:117–125
Tanabe A, Taketani S, Endo-Ichikawa Y, Tokunaga R, Ogawa Y, Hiramoto M (2000) Analysis of the candidate genes responsible for non-syndromic cleft lip and palate in Japanese people. Clin Sci (Lond) 99:105–111
van den Oord EJ, Vermunt JK (2000) Testing for linkage disequilibrium, maternal effects, and imprinting with (In)complete case-parents triads, by use of the computer program LEM. Am J Hum Genet 66:335–338
Vermunt JK (1997) LEM: a general program for the analysis of categorical data. Tilburg University, Tilburg
Vieira AR, Orioli IM, Castilla EE, Cooper ME, Marazita ML, Murray JC (2003) MSX1 and TGFB3 contribute to clefting in South America. J Dent Res 82:289–292
Weinberg CR, Wilcox AJ, Lie RT (1998) A log-linear approach to case-parent-triad data: assessing effects of disease genes that act either directly or through maternal effects and that may be subject to parental imprinting. Am J Hum Genet 62:969–978
Weinberg CR (1999) Methods for detection of parent-of-origin effects in genetic studies of case-parents triads. Am J Hum Genet 65:229–235
Weir BS (1996) Genetic data analysis II. Sinauer, Sunderland
Acknowledgments
We thank all participants for consenting to the study and providing blood samples. We are grateful to Birgit Kroschel-Lang, Charlotte Opitz, Michael Krimmel, and the German Support Group for People with Cleft Lip and/or Palate (Selbsthilfevereinigung für Lippen-Gaumen-Fehlbildungen e.V. Deutschland) for kind support in contacting patients. The authors also thank Susanne Raeder for skilful help with laboratory work. This study was supported by a grant from the Deutsche Forschungsgemeinschaft (DFG) to EM and FJK (FOR 423 Genetic Epidemiology and Medical Genetics of Complex Diseases, Project N1).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Reutter, H., Birnbaum, S., Mende, M. et al. TGFB3 displays parent-of-origin effects among central Europeans with nonsyndromic cleft lip and palate. J Hum Genet 53, 656–661 (2008). https://doi.org/10.1007/s10038-008-0296-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-008-0296-9
Keywords
This article is cited by
-
Genome-wide analysis of parent-of-origin effects in non-syndromic orofacial clefts
European Journal of Human Genetics (2014)
-
Intra-amniotic Transient Transduction of the Periderm With a Viral Vector Encoding TGFβ3 Prevents Cleft Palate in Tgfβ3−/− Mouse Embryos
Molecular Therapy (2013)
-
Contribution of MSX1 variants to the risk of non-syndromic cleft lip and palate in a Malay population
Journal of Human Genetics (2011)
-
Maternal transmission effects of the PAX genes among cleft case–parent trios from four populations
European Journal of Human Genetics (2009)
