
Abstract
Panic disorder (PD) is an anxiety disorder characterized by panic attacks and anticipatory anxiety. Although a number of association studies have been conducted, no gene has been identified as a susceptibility locus. In this study, we conducted a genome-wide association study of PD in 200 Japanese patients and the same number of controls, using the GeneChip Human Mapping 500 K Array Set. Genotypes were determined using the Bayesian Robust Linear Model with Mahalanobis (BRLMM) genotype calling algorithm. The genotype data were data-cleaned using criteria for SNP call rate (⩾95%), Hardy–Weinberg equilibrium (P⩾0.1%) and minor allele frequency (⩾5%). The significance level of the allele P-value was set at 1.0 × 10−6, to make false discovery rate (FDR) <0.05. As a result, seven SNPs were significantly associated with PD, which were located in or adjacent to genes including PKP1, PLEKHG1, TMEM16B, CALCOCO1, SDK2 and CLU (or APO-J). Studies with other samples are required to confirm the results.
Similar content being viewed by others

Introduction
Panic disorder (PD) is an anxiety disorder characterized by panic attacks and anticipatory anxiety. A life-time prevalence of PD is 1–3% and the female/male ratio among the affected is 2:1.1 Family and twin studies have consistently indicated that genes play a role in the etiology of PD. Although a number of association studies have been conducted, no gene has been identified as a susceptibility locus.
So far, in association studies, the candidate-gene approach has been employed. Particular attention has been addressed to anxiety-related genes, such as HTR 1A, 2A,2, 3, 4 CCK,5, 6 ADORA2A,7 COMT8, 9, 10 and MAOA.11 Most studies have focused on genes related to neurotransmitter systems as mentioned above. However, these studies have produced conflicting results.
Genome-wide association studies (GWAS) have become feasible because of the recent development of a microarray system that enables the simultaneous genotyping of several hundred thousands of SNPs. GWAS has been a powerful approach to the identification of genes involved in common human diseases without selecting candidate genes. A joint GWAS has been undertaken in the British population, and it examined seven major complex diseases including bipolar disorder, coronary artery disease, Crohn's disease, rheumatoid arthritis and type 1 and 2 diabetes.12 Regarding PD, Erhardt et al. (2007) reported that they were conducting a GWAS in 260 patients with anxiety disorders (mostly with PD) and 260 healthy controls using the Illumina BeadArray technology, but their result has not been published. No other association study has thus far performed a genome-wide search for susceptible loci of PD. In this study, we conducted a GWAS of PD in the Japanese population using a 500 K SNP chip.
Subjects and methods
Subjects
Subjects comprised 200 patients with PD (64 male and 136 female patients; age, 39.5±9.4 years, mean±s.d.) and the same number of healthy control subjects (64 male and 136 female patients; age, 38.2±9.3 years). All patients and control subjects were ethnically Japanese. The patients were recruited from two clinics for anxiety disorder in Tokyo and Nagoya, Japan. Nagoya is in the central area of Japan, 300 km apart from Tokyo. The patients were diagnosed according to the Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM-IV),13 and confirmed using the Mini International Neuropsychiatric Interview (M.I.N.I.) and reviewing the medical records. Healthy control subjects were mostly hospital staff who were recruited in Tokyo, Nagoya and the adjacent areas. The controls received a short interview by one of the authors and filled out a questionnaire to exclude those with a clinical history of psychiatric illness. The objective of this study was explained clearly and written informed consent was obtained from all subjects. The study was approved by the Ethical Committee of the Faculty of Medicine, the University of Tokyo.
Genotyping
The genomic DNA was extracted from peripheral leukocytes by standard procedures. Genotyping was conducted using the GeneChip Human Mapping 500 K Array Set (Affymetrix, CA, USA). A total of 500 568 SNPs on the chip were genotyped. This array set consisted of two chips (StyI and NspI) with approximately 250 000 SNPs on each chip. Genomic DNA (250 ng) was digested with restriction enzyme (StyI or NspI for each chip) and processed according to the manufacturer's protocol. The genotyping calls were analyzed using the GCOS1.4 and GTYPE4.1 software packages that employed the Bayesian Robust Linear Model with Mahalanobis (BRLMM) genotype calling algorithm.14
Statistical analysis
Allele frequencies were compared between patients and controls, and deviations from the Hardy–Weinberg equilibrium were calculated using the χ2 test. Statistical analyses were conducted using software developed for the analysis of large amounts of genotype data (GeneChipViewer, Dynacom, Japan).
We removed one control subject for the ‘Sty’ chip and one patient both for the ‘Sty’ and the ‘Nsp’ chips because the concentration of PCR products did not reach the expected level in the genotyping process. After the genotyping was completed, data cleaning was conducted as follows: (1) SNPs with a call rate of <95% in patients or controls were excluded, and (2) SNPs with a P-value of <0.1% for Hardy–Weinberg's equilibrium in the controls were excluded. (3) Then, the criterion regarding minor allele frequency (mAF) was determined as follows: the ratios of the observed and expected (by chance) numbers of SNPs with several levels of allele P-values (1.0 × 10−2, 10−3 and 10−4) were evaluated among SNPs with mAF of ⩾1, 5, 10% in all samples according to Miyagawa et al.15 When the observed numbers of SNPs with P-values were considered to be increased too much at a certain mAF compared with the expected numbers, SNPs with that (or a lower) level of mAF may be excluded, because, in that case, the ‘significant SNPs’ could contain too many false-positive ones. SNPs on the X chromosome were data-cleaned and analyzed statistically by sex.
The significance level of the allele P-value was determined by monitoring the level of the false discovery rate (FDR),16 which is the proportion of false-positive SNPs among all positive (or false-positive plus true-positive) ones. Under the null hypothesis, P-values should be uniformly distributed between 0 and 1; FDR methods typically consider the actual distribution as a mixture of outcomes under the null (uniform distribution of P-values) and alternative (P-value distribution skewed toward zero) hypotheses. FDR was calculated using the Genstat statistical package (BioSS, Scotland, UK). To exclude as many false-positive associations as possible, we set the FDR at <0.05. Under these conditions, the significance level of allele P-value was estimated to be <1.8 × 10−6. Thus, we tightly set the significance level of the allele P-value at 1.0 × 10−6 in this study. For replication study, additional data at the level of the allele P-value <1.0 × 10−4 were also shown.
The statistical power of the sample was calculated on the basis of the published method.17, 18
Results
The overall call rates of the genotyping assay were adequate in both patients and controls: 97.3 and 97.3% in controls, 98.0 and 97.7% in patients for the ‘Sty’ and ‘Nsp’ chips, respectively. Data cleaning was conducted for 490 032 SNPs on autosomal chromosomes and for 10 536 SNPs on the X chromosome. Among those SNPs, 371 589 SNPs on autosomal chromosomes, 9879 SNPs on the X chromosome in male patients and 8448 on the X chromosome in female patients satisfied the data-cleaning criterion of the call rate ⩾95% in both patients and controls. Among these SNPs, 368 721 SNPs on autosomal chromosomes and 8324 on the X chromosome in female patients satisfied the criterion of Hardy–Weinberg's equilibrium in controls (P⩾0.1%).
Among them, 275 329 SNPs on autosomal chromosomes, 7102 on the X chromosome in male patients and 5921 on the X chromosome in female patients were with mAF ⩾1%, 253 903, 6129 and 5110 were with mAF ⩾5% and 219 349, 5265 and 4441 were with mAF ⩾10%, respectively. The ratios of the observed/expected number of SNPs on autosomal chromosomes with the allele P-values of 1 × 10−2, 10−3 and 10−4 were 1.53, 2.19 and 4.07 for the SNPs with mAF ⩾1%, 1.22, 1.47, 1.89 for those with mAF ⩾5%, and 1.19, 1.37 and 1.27 for those with mAF ⩾10%, respectively (Table 1). According to these data, we employed mAF ⩾5% in all samples, considering the balance of possible false-positive and false-negative results. As a result, 253 903 SNPs on autosomal chromosomes, 6129 on the X chromosome in male patients and 5110 on the X chromosome in female patients were included in the present analysis after data cleaning.
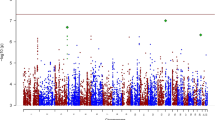
The log QQ P-value plot was shown after data cleaning using the criteria (SNP call rate ⩾95%, HWE P-value ⩾0.1% and mAF ⩾5%; Figure 1). Plots of P-values were close to the expected line (y=x). SNPs at the level of the allele P-value less than 0.01 are shown in Supplementary Table 1, where 4136 SNPs on autosomal chromosomes that were at the P levels in all subjects and 116 SNPs on the X chromosome in female patients were included.

Log quantile–quantile (QQ) P-value plot for the results after data cleaning. Cleaning criteria were single nucleotide polymorphism (SNP) call rate ⩾95%, Hardy–Weinberg equilibrium (HWE) P-value ⩾0.1% and minor allele frequency (mAF) ⩾5%.
Among these SNPs, seven were significantly associated with PD at a P-value less than 1.0 × 10−6. These SNPs included rs860554, rs9372078, rs17466684, rs12579350, rs941184, rs9302001 and rs3816995 (Table 2). Six of these SNPs were in genes, including PKP1, PLEKHG1, TMEM16B, CALCOCO1, SDK2 and one unknown gene. Another SNP (rs17466684) was within 1 kb of the CLU (also known as Apolipoprotein J) gene.
The strongest signal was with rs12579350 (allele P-value=3.7 × 10−9), which was located in the TMEM16B gene at 12p13 that may function as transporters for unidentified substrates. The interpretation of this signal may, however, require caution because the mAF of this SNP in controls was quite low (0.5%), whereas it was 9.9% in patients. The second strongest association was with rs860554 at 1q32, which was in PKP1 (P-value=4.6 × 10−8). The gene has an important role in the cytoskeleton/cell membrane interaction. The protein of PKP1, plackoglobin, acts as linker molecules at adherence junctions and desmosome at the plasma membrane. Among the other five SNPs, rs17466684 (P-value=6.8 × 10−7) at the chromosome 8p21 was located in the CLU (or Apolipoprotein J) gene. This gene may suppress beta-amyloid deposition in the brains of Alzheimer disease subjects. SNP rs3816995 (P-value=2.1 × 10−7) at the chromosome 17q24 was located in the SDK2 gene, which acts as determinants of lamina-specific synaptic connectivity. SNP rs941184 was located in the CALCOCO1 gene on 12q13. This protein binds to a highly conserved N-terminal domain and thus enhances transcriptional activation by a number of nuclear receptors.
As additional data, we show SNPs with the allele P-values between 1.0 × 10−6 and 1.0 × 10−4 in Table 3. Eleven SNPs were at the significance level of 1.0 × 10−6 ⩽P<1.0 × 10−5. Among the eleven, approximately a half (or six SNPs) was in or adjacent to genes, and the others were not. Thirty-one SNPs were at the significance level of 1.0 × 10−5 ⩽P<1.0 × 10−4, and a half of them (or 16 SNPs) were in or adjacent to genes, and the rest were not.
Discussion
We conducted a GWAS of panic disorder (PD) in Japanese, using a 500 K SNP chip. This may be the first report on GWAS of PD, to the best of our knowledge. We employed a set of data-cleaning criteria (SNP call rate ⩾95%, HWE P-value ⩾0.1% and mAF ⩾5%) for the data cleaning. The criterion of the mAF was determined considering the ratio of the observed and expected numbers of SNPs at the allele P-values of 1 × 10−2–10−4 in the comparison between patients and controls. The significance level for the allele P-value was set at P<1.0 × 10−6 by setting the false discovery rate (FDR) <0.05.
At the significance level of P<1.0 × 10−6, seven SNPs were associated with PD. Seven SNPs were within (n=6) or adjacent (n=1, within 1 kb) to genes, one of which is unknown. Among the seven SNPs, rs9302001, located on 13q32, is within the area of PAND1, which was associated with panic disorder/bladder syndrome.19 When mAFs in the controls are compared with those in the Japanese subjects in the HapMap database (http://www.ncbi.nlm.nih.gov/SNP/), the mAFs are similar in a portion of the SNPs including rs12579350 (mAF=0.5% in controls in this study and 0.0% in Japanese in the HapMap database, respectively) and rs860554 (mAF=4.9 and 6.4% respectively), whereas the mAFs are rather different in the rest of the SNPs. The results in them might be interpreted with caution. The SNP rs12579350 showed the strongest association (allele P-value=3.7 × 10−9) that should however also be interpreted with caution , because the mAF of this SNP was very low (<1%) in controls in contrast to close to 10% in patients.
As additional data, we showed SNPs at a significance level between 1.0 × 10−6 and 1.0 × 10−4 (Table 3). Forty-two SNPs were found at this level. It is interesting that fewer portions of the SNPs (22 of the 42) were located within the genes when this level of significance was employed, than at the level of P<1.0 × 10−6. The mAFs in the controls are quite similar to those in Japanese in the HapMap database in a portion of the SNPs including rs132617 in APOL3 and rs1383290 in DTNA, whereas the mAFs are rather different in another portion of the SNPs.
Several limitations may be acknowledged in this study. First, the sample size is limited (200 patients and 200 controls). The statistical power is estimated to be more than 0.9 for α=1.0 × 10−6, assuming that the prevalence of PD is 1.4% in the Japanese population, genotypic relative risk is 2.5 and minor allele frequency of the polymorphism is 0.3. For SNPs with less effects (or relative risks) or less allele frequencies, the sample size may be too small to detect the association with PD. Owing to the lack of power, several SNPs that are truly associated with PD might not be detected in this study. Candidate genes that were investigated in earlier association studies were not detected, including HTR 1A, 2A, ADORA2A, COMT and MAOA. This could be due to the lack of power in this study. Second, the number of SNPs in the statistical examination for the association with PD was decreased approximately to a half (or 254 000 SNPs) by the data cleaning. This might also affect the result of the genetic association to a degree, although the estimation is that 300 000 SNPs might capture most of the common genetic variations in a population.20 Finally, this study is a one-stage investigation, and therefore the SNPs with a significant association may be considered as candidates for future studies with other samples. The GWAS data will be available publicly in the Integrated Database (http://gwas.lifesciencedb.jp/).
References
Eaton, W. W., Kessler, R. C., Wittchen, H. U. & Magee, W. J. Panic and panic disorder in the United States. Am. J. Psychiatry 151, 413–420 (1994).
Inada, Y., Yoneda, H., Koh, J., Sakai, J., Himei, A ., Kinoshita, Y. et al. Positive association between panic disorder and polymorphism of the serotonin 2A receptor gene. Psychiatry Res. 118, 25–31 (2003).
Rothe, C., Gutknecht, L., Freitag, C., Tauber, R., Mossner, R ., Franke, P. et al. Association of a functional 1019C>G 5-HT1A receptor gene polymorphism with panic disorder with agoraphobia. Int. J. Neuropsychopharmacol. 7, 189–192 (2004).
Rothe, C., Koszycki, D., Bradwejn, J., King, N., De Luca, V ., Shaikh, S et al. Association study of serotonin-2A receptor gene polymorphism and panic disorder in patients from Canada and Germany. Neurosci. Lett. 363, 276–279 (2004).
Hattori, E., Yamada, K., Toyota, T., Yoshitsugu, K., Toru, M ., Shibuya, H et al. Association studies of the CT repeat polymorphism in the 5′ upstream region of the cholecystokinin B receptor gene with panic disorder and schizophrenia in Japanese subjects. Am. J. Med. Genet. 105, 779–782 (2001).
Ebihara, M., Ohba, H., Hattori, E., Yamada, K. & Yoshikawa, T. Transcriptional activities of cholecystokinin promoter haplotypes and their relevance to panic disorder susceptibility. Am. J. Med. Genet 118, 32–35 (2003).
Deckert, J., Nothen, M. M., Franke, P., Delmo, C., Fritze, J ., Knapp, M. et al. Systematic mutation screening and association study of the A1 and A2a adenosine receptor genes in panic disorder suggest a contribution of the A2a gene to the development of disease. Mol. Psychiatry 3, 81–85 (1998).
Hamilton, S. P., Slager, S. L., Heiman, G. A., Deng, Z., Haghighi, F ., Klein, D. F et al. Evidence for a susceptibility locus for panic disorder near the catechol-O-methyltransferase gene on chromosome 22. Biol. Psychiatry 51, 591–601 (2002).
Woo, J. M., Yoon, K. S. & Yu, B. H. Catechol O-methyltransferase genetic polymorphism in panic disorder. Am. J. Psychiatry 159, 1785–1787 (2002).
Woo, J. M., Yoon, K. S., Choi, Y. H., Oh, K. S., Lee, Y. S. & Yu, B. H. The association between panic disorder and the L/L genotype of catechol-O-methyltransferase. J. Psychiatr. Res. 38, 365–370 (2004).
Deckert, J., Catalano, M., Syagailo, Y. V., Bosi, M., Okladnova, O ., Di Bella, D. et al. Excess of high activity monoamine oxidase A gene promoter alleles in female patients with panic disorder. Hum. Mol. Genet. 8, 621–624 (1999).
Wellcome Trust Case Control Consortium. Genome-wide association study of 14 000 cases of seven common diseases and 3000 shared controls. Nature 447, 661–678 (2007).
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders 4th edn (American Psychiatric Association, Washington D.C., 1994).
Rabbee, N. & Speed, T. P. A genotype calling algorithm for affymetrix SNP arrays. Bioinformatics 22, 7–12 (2006).
Miyagawa, T., Nishida, N., Ohashi, J., Kimura, R., Fujimoto, A ., Kawashima, M. et al. Appropriate data cleaning methods for genome-wide association study. J. Hum. Genet. 53, 886–893 (2008).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B. 57, 289–300 (1995).
Ohashi, J., Yamamoto, S., Tsuchiya, N., Hatta, Y., Komata, T ., Matsushita, M. et al. Comparison of statistical power between 2 * 2 allele frequency and allele positivity tables in case-control studies of complex disease genes. Ann. Hum. Genet. 65, 197–206 (2001).
Ohashi, J. & Tokunaga, K. The expected power of genome-wide linkage disequilibrium testing using single nucleotide polymorphism markers for detecting a low-frequency disease variant. Ann. Hum. Genet. 66, 297–306 (2002).
Hamilton, S. P., Fyer, A. J., Durner, M., Heiman, G. A., Baisre de Leon, A ., Hodge, S. E. et al. Further genetic evidence for a panic disorder syndrome mapping to chromosome 13q. Proc. Natl Acad. Sci. USA 100, 2550–2555 (2003).
Balding, D. J. A tutorial on statistical methods for population association studies. Nat. Rev. Genet. 7, 781–791 (2006).
Acknowledgements
This study was supported by a Grant-in-Aid for Scientific Research on Priority Areas ‘Comprehensive Genomics’ and ‘Applied Genomics’ (no. 17019029) from the Ministry of Education, Culture, Sports, Science and Technology of Japan. We thank Ms Yoshimi Ishibashi and Ms Yuko Ogasawara for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website (http://www.nature.com/jhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Otowa, T., Yoshida, E., Sugaya, N. et al. Genome-wide association study of panic disorder in the Japanese population. J Hum Genet 54, 122–126 (2009). https://doi.org/10.1038/jhg.2008.17
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2008.17
Keywords
This article is cited by
-
Discovery and replication of blood-based proteomic signature of PTSD in 9/11 responders
Translational Psychiatry (2023)
-
Alterations in microRNA of extracellular vesicles associated with major depression, attention-deficit/hyperactivity and anxiety disorders in adolescents
Translational Psychiatry (2023)
-
Epistasis regulates the developmental stability of the mouse craniofacial shape
Heredity (2019)
-
Epigenome-wide association study of DNA methylation in panic disorder
Clinical Epigenetics (2017)
-
Meta-analysis of genome-wide SNP- and pathway-based associations for facets of neuroticism
Journal of Human Genetics (2017)
