
Abstract
The dystrophin gene, which is mutated in Duchenne and Becker muscular dystrophy, is characterized by its extremely large introns. Seven cryptic exons from the intronic sequences of the dystrophin gene have been shown to be inserted into the processed mRNA. In this study, we have cloned seven novel cryptic exons embedded in dystrophin introns that were amplified from dystrophin mRNA isolated from lymphocytes. All of these sequences, which ranged in size from 27 to 151 bp, were found to be cryptic exons because they were completely homologous to intronic sequences (introns 1, 18, 29, 63, 67, and 77), and possessed consensus sequences for branch points, splice acceptor sites, and splice donor sites. Compared with the 77 authentic dystrophin exons, the 14 cryptic exons were characterized by (1) lower Shapiro’s splicing probability scores for the splice donor and acceptor sites; (2) smaller and larger densities of splicing enhancer and silencer motifs, respectively; (3) a longer distance between the putative branch site and the splice acceptor site; and (4) with one exception, the introduction of premature stop codons into their respective transcripts. These characteristics indicated that the cryptic exons were weaker than the authentic exons. Our results suggested that a mutation deep within an intron that changed these parameters could cause dystrophinopathy. The cryptic exons identified provide areas that should be examined for the detection of mutations in the dystrophin gene, and they may help us to understand the roles of large dystrophin introns.
Similar content being viewed by others


Introduction
The human dystrophin gene, which is defective in patients with Duchenne or Becker muscular dystrophy (DMD/BMD), is the largest human gene; this gene spans more than 3,000 kb on the X-chromosome and encodes a 14-kb transcript that consists of 79 exons (Ahn and Kunkel 1993; Nishio et al. 1994). Therefore, some of the introns in the dystrophin gene are exceptionally large, covering more than 100 kb (Den Dunnen et al. 1989). However, the physiological roles of these introns are largely unknown, although fragments have been reported to contain alternative promoters and promoter-specific exons (Klamut et al. 1990; Boyce et al. 1991; Gorecki et al. 1992; Byers et al. 1993; Lederfein et al. 1993; Lidov et al. 1995).
Splicing, a process that removes intron sequences from pre-mRNA, is highly regulated to ensure the introns are removed and an ordered array of exons is assembled in mature transcripts. Splicing relies on the correct identification of exons, which must be recognized within pre-mRNA despite being relatively short compared to intronic regions. It is known that the presence of well-defined cis-elements, namely the splice donor and acceptor sites and the branch point, are necessary but not sufficient to define intron–exon boundaries (Senapathy et al. 1990). Other splicing regulatory elements include splicing enhancers and silencers in either exon or intron sequences (Ladd and Cooper 2002; Pozzoli and Sironi 2005). In the normal splicing reaction, introns are spliced out after the formation of a lariat that joins the 5′ end of an intron to the branch point. Some controversy, however, remains regarding the mechanism for splicing out large introns. The resplicing theory states that a large intron is spliced out by repeatedly splicing out shorter fragments. Evidence in support of this theory comes from the splicing of the Ultrabithorax gene of Drosophila melanogaster (Hatton et al. 1998). In human genes, however, no evidence supporting this theory has been reported.
Intronic DNA frequently encodes potential exonic sequences called pseudoexons that are not recognized by the splicing machinery (Sun and Chasin 2000). A subpopulation of pseudoexons can become splicing competent after subtle nucleotide changes. Indeed, it has been reported that single base-pair mutations or microdeletions deep within intronic regions can define novel exons by altering pseudoexon sequences without creating novel splice sites (Metherell et al. 2001; Ishii et al. 2002; Pagani et al. 2002).
Part of the 190-kb intron 1 of the dystrophin gene was found to be inserted into dystrophin transcripts in lymphocytes and was named exon 1a (Fig. 1a) (Roberts et al. 1993). The physiological role of cryptic exon 1a, which is inserted between exons 1 and 2 and encodes an in-frame stop codon, remains uncharacterized. Subsequently, six additional cryptic exons (exons 2a, 2b, 2c-1, 2c-s, 3a-l, and 3a-s) were disclosed in introns in the 5′ region of the dystrophin gene (Fig. 1a) (Dwi Pramono et al. 2000; Suminaga et al. 2002; Tran et al. 2005; Ishibashi et al. 2006). Because all of these exons encode a stop codon, their protein-coding abilities were not clear. Recently, a number of noncoding RNAs that are transcribed from the human genome have been cloned, which has shed new light on noncoding RNA (Carninci et al. 2005; Claverie 2005). In this study, we have identified seven novel cryptic dystrophin exons, and the characteristics of all 14 cryptic exons were compared with those of the 77 authentic dystrophin exons.

Cryptic exons in the dystrophin gene. Seven previously described cryptic exons and seven novel cryptic exons are schematically described in panels a and b, respectively. Boxes and lines represent exons and introns, respectively. Dotted and shaded boxes represent known and novel exons, respectively. Numbers in the boxes indicate the exon numbers. Numbers between the boxes in b indicate distances from the known exons
Cases and methods
Cases
More than 400 DMD patients from all over Japan were referred to the clinic for DMD patients at the Kobe University Hospital (Kobe, Japan). In clinically diagnosed cases of DMD, we attempted to identify the mutation in the dystrophin gene that was responsible. Genomic DNA was first analyzed by either conventional PCR amplification of the 19 deletion-prone exons of the dystrophin gene (Chamberlain et al. 1988; Beggs et al. 1990) or Southern blot analysis using HindIII-digested genomic DNA and amplified dystrophin cDNA as a probe (Mitsubishi Kagaku BCL Co., Tokyo, Japan) (Koenig et al. 1987). Then the 79 exons of the dystrophin gene were PCR amplified and directly sequenced.
In the cases that did not have any recognizable mutations in the analyzed genomic DNA, dystrophin mRNA expressed in lymphocytes was analyzed as described below. In some cases, dystrophin mRNA in lymphocytes was analyzed to clarify the molecular pathogenesis of the dystrophinopathy (Barbieri et al. 1996; Shiga et al. 1997; Adachi et al. 2003; Thi Tran et al. 2005). Consent for this study was obtained from the patient’s parents, and the ethics committee of the Kobe University Graduate School of Medicine approved the study.
Analysis of dystrophin mRNA
Total RNA was isolated from the peripheral lymphocytes of DMD patients and a control subject. Total RNA from human heart, lung, prostate, and skeletal muscle were purchased from BD Biosciences (San Jose, CA, USA). cDNA was synthesized as described previously (Matsuo et al. 1991). The entire dystrophin cDNA was analyzed by amplifying ten separate fragments using reverse-transcription (RT)-nested PCRs (Roberts et al. 1991). To identify splicing errors, a second PCR amplification of exons 1 to 11, 18 to 21, 25 to 30, 61 to 64, 66 to 72, or 70 to 79 was performed using primers (Hokkaido System Science Co., Sapporo, Japan) designed from the respective exons (Table 1a). PCR reactions (20 μl) contained 1 μl of cDNA, 2 μl of 10× Ex Taq buffer, 1 U of Ex Taq polymerase (Takara Bio Inc., Kyoto, Japan), 200 nM for each primer, and 250 mM dNTPs. The first amplification was performed using an outer set of primers and 30 cycles of 94 °C for 1 min, 58 °C for 1 min, and 72 °C for 3 min. A 1-μl sample of this reaction was then subjected to a second amplification using an inner set of primers and 30 cycles of 94 °C for 30 s, 60 °C for 30 s, and 72 °C for 1 min. A 5-μl sample of each PCR reaction was analyzed on a 2% NuSieve agarose gel containing 0.2 mg/ml ethidium bromide prior to photography.
For DNA sequencing, amplified products were purified, subcloned into the pT7 vector (Novagen, Madison, WI, USA), and the inserted DNA was sequenced with a Big Dye terminator cycle sequencing kit (Amersham Biosciences, Piscataway, NJ, USA) using an automatic DNA sequencer (ABI PRISM model 310, Applied Biosystems, Foster City, CA, USA), as described previously (Surono et al. 1997).
Analysis of genomic DNA
In order to identify genomic nucleotide changes that enhanced the incorporation of an intron fragment into the spliced mRNA, the cryptic exon and its flanking regions were examined by PCR amplification and direct sequencing. Cryptic exon-encompassing regions were PCR-amplified using primers designed from the flanking intron sequences (Table 1b); the conditions used have been described previously (Matsuo et al. 1990). The amplified product was directly sequenced by the method described above.
Characterization of cryptic exons
Shapiro’s splicing probability scores for the splice acceptor and donor sites were calculated for the 5′ and 3′ ends of the inserted sequences, respectively (Shapiro and Senapathy 1987). The splicing enhancer and silencer sequences were analyzed using the ACESCAN2 web server (http://genes.mit.edu/acescan2/index.html), which is an online tool for identifying candidate cis-elements in the splicing of mammalian exons. The densities of the enhancer or silencer motifs were obtained by counting the number of elements in each 100 bp of genomic sequence. Putative splicing enhancer and silencer motifs were predicted and counted.
To assess the significance of the values, the nonparametric Mann–Whitney rank test was employed as described previously (Roca et al. 2003).
Results
Dystrophin mRNA expressed in the lymphocytes of more than hundred DMD/BMD patients was analyzed by RT-nested PCR amplification to identify mutations in the dystrophin gene or to enhance molecular understanding of the dystrophinopathy. In some cases, subcloning sequencing of the amplified products disclosed some ambiguous clones, leading to the identification of cryptic exons. For example, the dystrophin mRNA in the lymphocytes of one DMD patient carrying a single G to A conversion at the first nucleotide of intron 20 (c.2622 + 1G>A), which destroyed a splice donor site, was examined for secondary splicing errors. When a fragment extending from exon 18 to 21 was amplified, two amplified products (one major and one minor) were obtained (Fig. 2). Subcloning sequencing of the major, normal-sized product disclosed a normal exon content. The size of exon 20, however, was shortened to 222 bp, because a cryptic splice donor site in exon 20 was activated due to inactivation of the authentic donor site, leading to a 20-bp deletion at the 3′ end of exon 20. On the other hand, the larger, minor product was composed of exons 18 to 21 and an additional 132-bp sequence between exons 18 and 19 (Fig. 2). A BLAST search with the 132-bp sequence revealed the sequence was derived from intron 18 of the dystrophin gene (nt 30314022-30313891 of GenBank NT_011757). The inserted 132-bp sequence was located 3.8 kb downstream from exon 18 and 12.2 kb upstream of exon 19 (Fig. 1). AG and GT dinucleotides, which are absolutely conserved at splice acceptor and donor sites of all introns, were identified in the genomic sequence immediately adjacent to the 5′ and 3′ ends of the 132-bp sequence, respectively. The Shapiro’s splicing probability scores for the splice acceptor and donor sites were 0.73 and 0.64, respectively (Shapiro and Senapathy 1987). Furthermore, the branch-point consensus sequence TGCTCAT was identified 38 bp upstream of the inserted sequence (Table 2). Seven splicing enhancer sequences and three splicing silencer sequences within the 132-bp sequence were identified (Table 2). Because the inserted sequence exhibited all of the characteristics typical of a genomic exon and was inserted between authentic dystrophin exons, we refer to this sequence as dystrophin exon 18a.

Identification of cryptic exon 18a. a RT-nested PCR amplification of a fragment containing exons 18 to 21 from the lymphocyte of a DMD patient carrying a single nucleotide conversion of the first nucleotide of intron 20 (c.2622 + 1G>A). Two products were visualized on the gel; one was barely visible, whereas the other one was clearly visible (P). One amplified product was obtained from the control sample (C). A schematic representation of the exonic organization in the amplified fragments is shown to the right of the products. The black boxes correspond to the 20-bp deletion in exon 20. C and P refer to the control sample and the index case, respectively. b The sequences at the junctions between the 132-bp inserted sequence and the flanking authentic exons and the junction between exon 20 and 21 are shown. The terminal sequence of exon 18 (CAAT) is joined to the 5′ end of the 132-bp inserted sequence (TGGA), whereas the 3′ end of the insert (CCAG) is joined to the 5′ end of the sequence of exon 19 (GCCA) (upper panel). In the junction between exon 20 and 21, 20 bp of the 3′ end of exon 20 are deleted (upper panel). The normal junction between exons 18 and 19 is shown (lower panel)
We then looked for a genomic mutation that could have caused the activation of exon 18a by sequencing the intronic region near the inserted 132-bp sequence. Because the genomic sequence was completely normal, however, the activation of exon 18a was attributed to a remote effect induced by a nucleotide change at some distant location (Suminaga et al. 2002). When the protein-coding ability of exon 18a was examined by analyzing the translational reading frame of the resulting dystrophin mRNA, a premature stop codon was identified in the 26th codon of exon 18a. Therefore, it is unlikely that transcripts containing exon 18a would code for the translation of a novel dystrophin protein isoform.
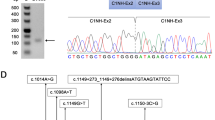
Subcloning sequencing of products amplified with RT-nested PCRs from dystrophin mRNA isolated from lymphocytes subsequently disclosed five unknown sequences that were inserted into at least one sequencing clone (Fig. 3). When a fragment containing exons 1 to 11 was amplified, one clone contained a 105-bp insertion between exons 1 and 2 together with a deletion of exons 3 to 6 (Table 2). When a fragment spanning from exon 25 to 30 was amplified, a 45-bp insertion between exons 29 and 30 was identified in one clone that also carried a single G nucleotide deletion in exon 27 (c.3613del G) (Table 2). In one clone containing an amplified fragment spanning from exons 61 to 64, a 75-bp insertion between exons 63 and 64 was identified, although no mutation was found yet in the dystrophin gene (Table 2). Remarkably, in one DMD patient in whom no mutation in the dystrophin gene had been identified thus far, two insertions were disclosed: a 51-bp insertion between exons 67 and 68 and a 151-bp insertion between exons 77 and 78 were cloned when fragments encompassing exons 66 to 72 and exons 70 to 79 were examined, respectively. All of the inserted sequences were completely homologous to the respective intron sequences (Fig. 2), and AG and GT dinucleotides immediately flanked the 5′ and 3′ ends of each insertion, respectively (Table 2). Furthermore, a consensus sequence for a branch point was identified upstream of each insertion (Table 2). Therefore, these insertions were named exons 1b, 29a, 63a, 67a, and 77a. Of these insertions, only exon 67a did not disrupt the open reading frame of the dystrophin mRNA.

Sequencing results for five of the novel cryptic exons. Sequences at the junctions of the inserted sequences and the flanking authentic exons are shown. Numbers over the sequencing charts indicate the nucleotide positions of the cryptic exons in the sequence of Genebank NT_011757
The disclosure of exon 1b in addition to exon 1a in intron 1 suggested that these cryptic exons have physiological roles. A fragment encompassing exons 1b and 1a was examined using cDNA prepared from normal heart, lung, prostate, and skeletal muscle (Fig. 4). The amount of the PCR-amplified fragment obtained from heart and skeletal muscle was sufficient for further analysis, whereas this was not the case for the samples from lung and prostate. Subcloning sequencing of the product disclosed two clones: one consisted of exons 1b and 1a and the other contained an unknown 27-bp insertion between exon 1b and 1a. The inserted sequence was completely homologous to a region in intron 1 (nt 30885727 to 30885701 of Genebank NT_011757). In the genomic sequence, we identified AG and GT dinucleotides immediately adjacent to the 5′ and 3′ end of the insertion, respectively, in addition to a branch point consensus sequence upstream of the insertion; therefore, we named the insertion exon 1c (Table 2). Both of the cloned sequences, however, encoded stop codons in every frame and were not expected to code for proteins. BLAST homology searches in Genbank with the full-length clones disclosed no homologous sequences.

Cloning of cryptic exon 1c. a RT-PCR products are shown. Total RNA from four sources was analyzed by RT-PCR amplification of a fragment encompassing two cryptic exons (exon 1b and 1a). One product was amplified from heart and skeletal muscle, but not from lung or prostate. A schematic representation of the exonic organization in the amplified fragments is shown to the right of the products. b Sequences at the junctions of the exons are shown. Subcloning sequencing of the amplified products disclosed the presence of two different clones. In one clone, the 3′ end of exon 1b was directly joined to the 5′ end of exon 1a (lower panel), whereas the 27-bp cryptic exon was inserted between exons 1b and 1a in the other clone
To date, 14 cryptic exons have been identified in introns 1, 2, 3, 18, 29, 63, 67, and 77. In order to identify elements that differentiate cryptic exons from authentic exons, the characteristics of the 14 cryptic and 77 authentic exons were examined. The length of the cryptic exons ranged from 27 to 357 bp (mean 123 ± 82 bp), whereas that of the authentic exons ranged from 32 to 269 bp (mean 143 ± 49 bp); the mean length of the cryptic exons was smaller than that of the authentic exons. The strengths of the splicing acceptor and donor sites were calculated using the Shapiro’s splicing probability score (Fig. 5). In a plot of the scores for the acceptor versus the donor sites, many of the cryptic exons were plotted in the area where the two splicing scores were low, whereas many of the authentic exons were plotted in the area where the two scores were high. The Shapiro’s scores for the splice donor sites of the cryptic exons ranged from 0.61 to 0.92 (mean 0.78 ± 0.07), whereas those of the authentic exons ranged from 0.67 to 1 (mean 0.83 ± 0.07) (Sironi et al. 2001); the scores for cryptic exons were significantly lower than those for the authentic exons (Fig. 5). In addition, the mean probability score for the splice acceptor sites of the cryptic exons was significantly lower than that for the splice acceptor sites of the authentic exons (Fig. 5; mean 0.79 ± 0.12 vs. 0.87 ± 0.09, respectively).

Strength of the splice sites. a A graphical representation of the Shapiro’s splicing probability scores for the splice donor and acceptor sites. Black and open boxes represent the cryptic and authentic exons, respectively. b Comparison of the Shapiro’s splicing probability scores. Bars indicate the mean values of the splice donor (upper) and acceptor (lower) sites. Shaded and dotted bars represent authentic and cryptic exons, respectively
Because the incorporation of exons into mRNA is controlled by splicing enhancer and/or silencer sequences in exons, the densities of the splicing enhancer and silencer sequences in each exon were analyzed (Fig. 6a,b). In a plot of the density of splicing enhancers versus the density of splicing silencers, most of the cryptic exons were plotted in the area corresponding to a larger density of splicing silencers compared to the density of enhancers. In contrast, authentic exons had a smaller density of splicing silencers and a larger density of enhancers. The mean density of splicing enhancers in the cryptic exons was smaller than that in authentic exons (9.8 vs. 16.2 per 100 bp of genomic sequence), and the mean density of splicing silencers was significantly larger in cryptic exons than in authentic exons (3.9 vs. 2.1 per 100 bp of genomic sequence) (Fig. 6b). This suggested that the spliceosome recognized the authentic exons better than the cryptic exons.

Examination of genomic sequences. a A graphical representation of the densities of the splicing enhancer and silencer motifs. Black and open boxes represent cryptic and authentic exons, respectively. b Comparison of splicing enhancer and silencer densities. Bars indicate mean values of the densities of splicing enhancer (left) and silencer (right) motifs. Shaded and dotted bars represent authentic and cryptic exons, respectively. c, d Comparison of length and the pyrimidine content of the sequences between the putative branch point and the splice acceptor site. The length (c) and pyrimidine content (d) of the sequence between the putative branch point and the splice acceptor site are shown. Shaded and dotted bars represent authentic and cryptic exons, respectively
The distance between the splice acceptor site and the putative branch point was examined and compared in the two types of exons. The mean distance between the acceptor site and the branch point for the cryptic exons was 64 bp (range 20–116 bp), whereas that for the authentic exons was 44 bp (range 16–123 bp); the mean distance was significantly longer for the cryptic exons (Fig. 6c), which indicated that the cryptic exons were weaker than the authentic exons. Additionally, the pyrimidine nucleotide content, which is an indication of the strength of a splice site, was examined for the nucleotide sequences between the branch points and the splicing acceptor sites. For the cryptic exons, the mean pyrimidine content was 67% (range: 50–82%), whereas it was 63% (range 37–89%) for the authentic exons; this difference was not significant (Fig. 6c).
The introns in the human dystrophin gene have increased in size through the acquisition of new elements into the introns (McNaughton et al. 1997). To examine whether the cryptic exons were products of exonization from retrotransposable elements, homology searches were done with each cryptic exon (http://www.repeatmasker.org/cgi-bin/WEBRepeatMasker). As a result, exon 1a and 2b were found within ER-V class I and Alu sequences, respectively. The other cryptic exons, however, did not contain repetitive elements, and the mechanism for exonization was consequently more difficult to establish.
Resplicing has been reported to explain the mechanism by which large introns are spliced out of pre-mRNA (Hatton et al. 1998; Burnette et al. 2005). Because the dystrophin gene has extremely large introns, it is possible that splicing proceeds via a resplicing mechanism that uses AGGT tetranucleotides as splice acceptor and donor sites. The sequences of the cryptic exons were examined for GT and AG dinucleotides at the 5′ and 3′ ends of the inserts, respectively. Among the 14 cryptic exons, only exon 2a contained a GT dinucleotide at its 5′ end, which made the junction between exon 2 and 2a an AGGT tetranucleotide. At this tetranucleotide, the Shapiro’s splicing probability scores for the splice acceptor and donor sites were 0.75 and 0.78, respectively, making this a good candidate for a resplicing site. At the 3′ end of exon 2a, however, there was a GT but not an AG dinucleotide; therefore, this position was not suitable as a resplicing substrate. Therefore, it is unlikely that the cryptic exons were intermediate products of resplicing.
In conclusion, compared to the authentic exons, the cryptic exons were shorter, and had lower splicing acceptor and donor scores, smaller densities of splicing enhancers, larger densities of splicing silencers, and branch points that were farther away from the splicing acceptor sites. All of these characteristics suggest that the cryptic exons were weaker than the authentic exons.
Discussion
The extremely large introns in the dystrophin gene suggest that a specific mechanism is required for the processing of dystrophin mRNA (Surono et al. 1997, 1999). Seven cryptic exons have been identified in dystrophin introns due to the incorporation of intron fragments into dystrophin mRNA (Fig. 1) (Roberts et al. 1993; Dwi Pramono et al. 2000; Suminaga et al. 2002; Tran et al. 2005; Ishibashi et al. 2006). In this study, seven additional fragments of introns of the dystrophin gene were shown to be inserted into dystrophin mRNA (Table 2). These cryptic exons are smaller and have weak splice site signals, which is commonly observed for alternatively spliced exons (Fig. 5) (Baek and Green 2005; Yeo et al. 2005).
It has been estimated that there is one pseudoexon for every kilobase of intron (Sironi et al. 2004). Therefore, more than 100 pseudoexons should be present in the 190-kb intron 1 of the dystrophin gene. In this report, we identified two additional cryptic exons in intron 1: exon 1b and exon 1c (Fig. 1). It is likely that other fragments of intron 1 will be identified as cryptic exons. In intron 1, one candidate sequence (121 nt from 56074 to 55954 of Genbank AL049643) was identified 40 kb upstream from exon 1b with a computer-based search for exonic sequences. No incorporation of this sequence into dystrophin mRNA, however, was observed (data not shown). This suggests that not all sequences that have exonic characteristics are active, and that there are specific mechanisms to allow the spliceosome to recognize exon sequences.
Intron 29 is known to encode the promoter and the first exon (exon R1) of R-dystrophin, one of the dystrophin isoforms expressed in retina (Pillers et al. 1993). Exon 29a is different from the other cryptic exons in the dystrophin gene, because it shares a common splicing donor site with exon R1 of the R-dystrophin transcript (Fig. 1). Though exon R1 is 95 bp long, exon 29 uses a cryptic splice acceptor site located 45 bp upstream from the 3′ end of exon R1 (Fig. 1). Considering that the splice donor site of exon R1 is activated in the retina (Pillers et al. 1993), exon 29a may be specifically expressed in the retina, which should be addressed in future studies.
It is known that mRNA encoding a premature stop codon can be subjected to degradation by the nonsense-mediated mRNA decay mechanism (Maquat 2004). Because our results indicated that the cryptic exons eluded this surveillance (Hillman et al. 2004), the transcripts containing the cryptic exons are suggested to have physiological roles. Five potential explanations could account for the presence of these cryptic exons in dystrophin mRNA. First, they could be aberrant splicing products. All of the cryptic exons except exon 1c were detected in dystrophin mRNA isolated from lymphocytes where the dystrophin gene is illegitimately expressed. Therefore, we believe that the splicing of dystrophin pre-mRNA is not as strictly controlled in lymphocytes as it is in skeletal muscle, which allows the splicing machinery to recognize cryptic exons. Second, these exons may constitute untranslated regions of dystrophin mRNA variants. This possibility is based on the fact that nine of the 14 cryptic exons were identified in introns located upstream of exon 8, which has been reported to include an alternative translation start site (Malhotra et al. 1988; Gangopadhyay et al. 1992). Therefore, the nine cryptic exons upstream of exon 8 could make up part of the 5′-untranslated region of a dystrophin mRNA variant. In fact, an alternative exon has been identified in the 5′-untranslated region the P-dystrophin transcript (Holder et al. 1996). Complex patterns of alternative splicing of the 5′ region of the dystrophin gene have been reported and are considered to be a factor that can explain some cases in which there is a discrepancy between the phenotype and molecular genotype (Chelly et al. 1991; Reiss and Rininsland 1994; Torelli and Muntoni 1996; Surono et al. 1997).
A third possibility is that the cryptic exons are intermediate products of a resplicing process. Whether one-step splicing is used to remove the large intron sequences has long been debated (Hatton et al. 1998). The resplicing theory states that repetitive splicing of small fragments allows the removal of large introns (Hatton et al. 1998). Because the dystrophin gene has large introns, the identified cryptic exons may be intermediate products of resplicing that uses AGGT tetranucleotides as splice donor and acceptor sites. Only exon 2a, however, had a GT dinucleotide at its 5′ end, and exon 2a did not have an AG dinucleotide at its 3′ end, preventing the formation of an AGGT tetranucleotide with the downstream exon sequence. In addition, the sizes of the introns containing the cryptic exons are 189, 170, 4.8, 16, 26, 38, 21, and 7.4 kb for introns 1, 2, 3, 18, 29, 63, 67, and 77, respectively. Therefore, it is unlikely that all of these cryptic exons are resplicing intermediate products.
The fourth possibility is that these cryptic exons are part of noncoding RNAs. It has been suggested that noncoding transcripts play important roles in the downregulation of gene expression (Hillman et al. 2004; Lareau et al. 2004). Recently, a large number of noncoding RNAs were identified in humans (Carninci et al. 2005), and it is likely that there are many noncoding RNAs that are yet to be uncovered. Therefore, the dystrophin cryptic exons may be parts of noncoding RNAs that regulate gene expression.
Finally, the exons may be intermediates of exonization. Recent data suggest that newly created exons result in an expansion of proteomic diversity (Lev-Maor et al. 2003; Sorek et al. 2004). This is probably accomplished through a multistep process in which the novel exon is initially included in a minority of the transcripts (due to weak splicing signals) and is thereby allowed to evolve without compromising the original gene function. Repeated genomic elements have been reported to be exonized (Lev-Maor et al. 2003; Sorek et al. 2004). Sequence analysis of the 14 cryptic exons disclosed some homology in exons 1a, 1b, and 2b to the repeat sequences. Future studies are required to address this possibility.
In the dystrophin gene a single nucleotide change located at deep intron has been reported to cause dystrophinopathy; creation or reinforcement of a splicing donor site consensus sequence has been reported in introns 9, 25, 60, and 62 (Ikezawa et al. 1998; Tuffery-Giraud et al. 2003) or 62 (Beroud et al. 2004), respectively. In addition, creation or reinforcement of a splice acceptor site consensus sequence has been reported in introns 1 and 2 (Yagi et al. 2003; Beroud et al. 2004) or 25 (Tuffery-Giraud et al. 2003), respectively. Until now any single nucleotide change affecting exonic splicing silencer or enhancer of cryptic exons of the dystrophin gene has not been reported. Pseudoexon insertions, however, have been reported in the ATM gene (Eng et al. 2004). Further studies may disclose mutations deep in introns that cause dystrophinopathy. In addition, our identification of these cryptic exons may shed light on the diverse functions of dystrophin.
References
Adachi K, Takeshima Y, Wada H, Yagi M, Nakamura H, Matsuo M (2003) Heterogous dystrophin mRNAs produced by a novel splice acceptor site mutation in intermediate dystrophinopathy. Pediatr Res 53:125–131
Ahn AH, Kunkel LM (1993) The structural and functional diversity of dystrophin. Nat Genet 3:283–291
Baek D, Green P (2005) Sequence conservation, relative isoform frequencies, and nonsense-mediated decay in evolutionarily conserved alternative splicing. Proc Natl Acad Sci USA 102:12813–12818
Barbieri AM, Soriani N, Ferlini A, Michelato A, Ferrari M, Carrera P (1996) Seven novel additional small mutations and a new alternative splicing in the human dystrophin gene detected by heteroduplex analysis and restricted RT-PCR heteroduplex analysis of illegitimate transcripts. Eur J Hum Genet 4:183–187
Beggs AH, Koenig M, Boyce FM, Kunkel LM (1990) Detection of 98% of DMD/BMD gene deletions by polymerase chain reaction. Hum Genet 86:45–48
Beroud C, Carrie A, Beldjord C, Deburgrave N, Llense S, Carelle N, Peccate C, Cuisset JM, Pandit F, Carre-Pigeon F, Mayer M, Bellance R, Recan D, Chelly J, Kaplan JC, Leturcq F (2004) Dystrophinopathy caused by mid-intronic substitutions activating cryptic exons in the DMD gene. Neuromuscul Disord 14:10–18
Boyce FM, Beggs AH, Feener C, Kunkel LM (1991) Dystrophin is transcribed in brain from a distant upstream promoter. Proc Natl Acad Sci USA 88:1276–1280
Burnette JM, Miyamoto-Sato E, Schaub MA, Conklin J, Lopez AJ (2005) Subdivision of large introns in Drosophila by recursive splicing at nonexonic elements. Genetics 170:661–674
Byers TJ, Lidov HG, Kunkel LM (1993) An alternative dystrophin transcript specific to peripheral nerve. Nat Genet 4:77–81
Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R et al. (2005) The transcriptional landscape of the mammalian genome. Science 309:1559–1563
Chamberlain JS, Gibbs RA, Ranier JE, Nguyen PN, Caskey CT (1988) Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucleic Acids Res 16:11141–11156
Chelly J, Gilgenkrantz H, Hugnot JP, Hamard G, Lambert M, Recan D, Akli S, Cometto M, Kahn A, Kaplan JC (1991) Illegitimate transcription. Application to the analysis of truncated transcripts of the dystrophin gene in nonmuscle cultured cells from Duchenne and Becker patients. J Clin Invest 88:1161–1166
Claverie JM (2005) Fewer genes, more noncoding RNA. Science 309:1529–1530
Den Dunnen JT, Grootscholten PM, Bakker E, Blonden LA, Ginjaar HB, Wapenaar MC, van Paassen HM, van Broeckhoven C, Pearson PL, van Ommen GJ (1989) Topography of the Duchenne muscular dystrophy (DMD) gene: FIGE and cDNA analysis of 194 cases reveals 115 deletions and 13 duplications. Am J Hum Genet 45:835–847
Dwi Pramono ZA, Takeshima Y, Surono A, Ishida T, Matsuo M (2000) A novel cryptic exon in intron 2 of the human dystrophin gene evolved from an intron by acquiring consensus sequences for splicing at different stages of anthropoid evolution. Biochem Biophys Res Commun 267:321–328
Eng L, Coutinho G, Nahas S, Yeo G, Tanouye R, Babaei M, Dork T, Burge C, Gatti RA (2004) Nonclassical splicing mutations in the coding and noncoding regions of the ATM Gene: maximum entropy estimates of splice junction strengths. Hum Mutat 23:67–76
Gangopadhyay SB, Sherratt TG, Heckmatt JZ, Dubowitz V, Miller G, Shokeir M, Ray PN, Strong PN, Worton RG (1992) Dystrophin in frameshift deletion patients with Becker muscular dystrophy. Am J Hum Genet 51:562–570
Gorecki DC, Monaco AP, Derry JMJ, Walker AP, Barnard EA, Barnard PJ (1992) Expression of four alternative dystrophin transcripts in brain regions regulated by different promoters. Hum Mol Genet 1:505–510
Hatton AR, Subramaniam V, Lopez AJ (1998) Generation of alternative Ultrabithorax isoforms and stepwise removal of a large intron by resplicing at exon–exon junctions. Mol Cell 2:787–796
Hillman RT, Green RE, Brenner SE (2004) An unappreciated role for RNA surveillance. Genome Biol 5:R8
Holder E, Maeda M, Bies RD (1996) Expression and regulation of the dystrophin purkinje promoter in human skeletal muscle, heart, and brain. Hum Genet 97:232–239
Ikezawa M, Minami N, Takahashi M, Goto Y, Miike T, Nonaka I (1998) Dystrophin gene analysis on 130 patients with Duchenne muscular dystrophy with a special reference to muscle mRNA analysis. Brain Dev 20:165–168
Ishibashi K, Takeshima Y, Yagi M, Nishiyama A, Matsuo M (2006) Novel cryptic exons identified in introns 2 and 3 of the human dystroiphin gene with duplication of exons 8–11. Kobe J Med Sci 52:61–75
Ishii S, Nakao S, Minamikawa-Tachino R, Desnick RJ, Fan JQ (2002) Alternative splicing in the alpha-galactosidase A gene: increased exon inclusion results in the Fabry cardiac phenotype. Am J Hum Genet 70:994–1002
Klamut HJ, Gangopadhyay SB, Worton RG, Ray PN (1990) Molecular and functional analysis of the muscle specific promoter region of the Duchenne muscular dystrophy gene. Mol Cell Biol 10:193–205
Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM (1987) Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 50:509–517
Ladd AN, Cooper TA (2002) Finding signals that regulate alternative splicing in the post-genomic era. Genome Biol 3:8.1–8.16
Lareau LF, Green RE, Bhatnagar RS, Brenner SE (2004) The evolving roles of alternative splicing. Curr Opin Struct Biol 14:273–282
Lederfein D, Yaffe D, Nudel U (1993) A housekeeping type promoter, located in the 3′ region of the Duchenne muscular dystrophy gene, controls the expression of Dp71, a major product of the gene. Hum Mol Genet 2:1883–1888
Lev-Maor G, Sorek R, Shomron N, Ast G (2003) The birth of an alternatively spliced exon: 3′ splice-site selection in Alu exons. Science 300:1288–1291
Lidov HG, Selig S, Kunkel LM (1995) Dp140: a novel 140 kDa CNS transcript from the dystrophin locus. Hum Mol Genet 4:329–335
Malhotra SB, Hart KA, Klamut HJ, Thomas NST, Bodrug E, Burghes AHM, Borrow M, Harper PS, Thompson W, Ray PN (1988) Frame-shift deletions in patients with Duchenne and Becker muscular dystrophy. Science 242:755–759
Maquat LE (2004) Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol 5:89–99
Matsuo M, Masumura T, Nakajima T, Kitoh Y, Takumi T, Nishio H, Koga J, Nakamura H (1990) A very small frame-shifting deletion within exon 19 of the Duchenne muscular dystrophy gene. Biochem Biophys Res Commun 170:963–967
Matsuo M, Masumura T, Nishio H, Nakajima T, Kitoh Y, Takumi T, Koga J, Nakamura H (1991) Exon skipping during splicing of dystrophin mRNA precursor due to an intraexon deletion in the dystrophin gene of Duchenne muscular dystrophy Kobe. J Clin Invest 87:2127–2131
McNaughton JC, Hughes G, Jones WA, Stockwell PA, Klamut HJ, Petersen GB (1997) The evolution of an intron: analysis of a long, deletion-prone intron in the human dystrophin gene. Genomics 40:294–304
Metherell LA, Akker SA, Munroe PB, Rose SJ, Caulfield M, Savage MO, Chew SL, Clark AJ (2001) Pseudoexon activation as a novel mechanism for disease resulting in atypical growth-hormone insensitivity. Am J Hum Genet 69:641–646
Nishio H, Takeshima Y, Narita N, Yanagawa H, Suzuki Y, Ishikawa Y, Minami R, Nakamura H, Matsuo M (1994) Identification of a novel first exon in the human dystrophin gene and of a new promoter located more than 500 kb upstream of the nearest known promoter. J Clin Invest 94:1037–1042
Pagani F, Buratti E, Stuani C, Bendix R, Dork T, Baralle FE (2002) A new type of mutation causes a splicing defect in ATM. Nature Genet 30:426–429
Pillers D-AM, Bulman DE, Welber RG, Sigesmund DA, Musarella MA, Powell BR, Murphey WH, Westall C, Panton C, Becker LE, Worton RG, Ray PN (1993) Dystrophin expression in the human retina is required for normal function as defined by electroretinography. Nature Genet 4:82–86
Pozzoli U, Sironi M (2005) Silencers regulate both constitutive and alternative splicing events in mammals. Cell Mol Life Sci 62:1579–1604
Reiss J, Rininsland F (1994) An explanation for the constitutive exon 9 cassette splicing of the DMD gene. Hum Mol Genet 3:295–298
Roberts RG, Barby TF, Manners E, Bobrow M, Bentley DR (1991) Direct detection of dystrophin gene rearrangements by analysis of dystrophin mRNA in peripheral blood lymphocytes. Am J Hum Genet 49:298–310
Roberts RG, Bentley DR, Bobrow M (1993) Infidelity in the structure of ectopic transcripts: a novel exon in lymphocyte dystrophin transcripts. Hum Mutat 2:293–299
Roca X, Sachidanandam R, Krainer AR (2003) Intrinsic differences between authentic and cryptic 5′ splice sites. Nucleic Acids Res 31:6321–6333
Senapathy P, Shapiro MB, Harris NL (1990) Splice junctions, branch point sites, and exons: sequence statistics, identification, and applications to genome project. Methods Enzymol 183:252–278
Shapiro MB, Senapathy P (1987) RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res 15:7155–7174
Shiga N, Takeshima Y, Sakamoto H, Inoue K, Yokota Y, Yokoyama M, Matsuo M (1997) Disruption of the splicing enhancer sequence within exon 27 of the dystrophin gene by a nonsense mutation induces partial skipping of the exon and is responsible for Becker muscular dystrophy. J Clin Invest 100:2204–2210
Sironi M, Menozzi G, Riva L, Cagliani R, Comi GP, Bresolin N, Giorda R, Pozzoli U (2004) Silencer elements as possible inhibitors of pseudoexon splicing. Nucleic Acids Res 32:1783–1791
Sironi M, Pozzoli U, Cagliani R, Comi GP, Bardoni A, Bresolin N (2001) Analysis of splicing parameters in the dystrophin gene: relevance for physiological and pathogenetic splicing mechanisms. Hum Genet 109:73–84
Sorek R, Lev-Maor G, Reznik M, Dagan T, Belinky F, Graur D, Ast G (2004) Minimal conditions for exonization of intronic sequences: 5′ splice site formation in alu exons. Mol Cell 14:221–231
Suminaga R, Takeshima Y, Adachi K, Yagi M, Nakamura H, Matsuo M (2002) A novel cryptic exon in intron 3 of the dystrophin gene was incorporated into dystrophin mRNA with a single nucleotide deletion in exon 5. J Hum Genet 47:196–201
Sun H, Chasin LA (2000) Multiple splicing defects in an intronic false exon. Mol Cell Biol 20:6414–6425
Surono A, Takeshima Y, Wibawa T, Ikezawa M, Nonaka I, Matsuo M (1999) Circular dystrophin RNAs consisting of exons that were skipped by alternative splicing. Hum Mol Genet 8:493–500
Surono A, Takeshima Y, Wibawa T, Pramono ZA, Matsuo M (1997) Six novel transcripts that remove a huge intron ranging from 250 to 800 kb are produced by alternative splicing of the 5′ region of the dystrophin gene in human skeletal muscle. Biochem Biophys Res Commun 239:895–899
Thi Tran HT, Takeshima Y, Surono A, Yagi M, Wada H, Matsuo M (2005) A G-to-A transition at the fifth position of intron 32 of the dystrophin gene inactivates a splice donor site both in vivo and in vitro. Mol Genet Metab 85:213–219
Torelli S, Muntoni F (1996) Alternative splicing of dystrophin exon 4 in normal human muscle. Hum Genet 97:521–523
Tran VK, Zhang Z, Yagi M, Nishiyama A, Habara Y, Takeshima Y, Matsuo M (2005) A novel cryptic exon identified in the 3′ region of intron 2 of the human dystrophin gene. J Hum Genet 50:425–433
Tuffery-Giraud S, Saquet C, Chambert S, Claustres M (2003) Pseudoexon activation in the DMD gene as a novel mechanism for Becker muscular dystrophy. Hum Mutat 21:608–614
Yagi M, Takeshima Y, Wada H, Nakamura H, Matsuo M (2003) Two alternative exons can result from activation of the cryptic splice acceptor site deep within intron 2 of the dystrophin gene in a patient with as yet asymptomatic dystrophinopathy. Hum Genet 112:164–170
Yeo GW, Van Nostrand E, Holste D, Poggio T, Burge CB (2005) Identification and analysis of alternative splicing events conserved in human and mouse. Proc Natl Acad Sci USA 102:2850–2855
Acknowledgments
We would like to thank Ms. A. Hosoda for her secretarial help. This work was supported by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science; Health and Labour Sciences Research Grants from the Ministry of Health, Labour and Welfare of Japan for research on psychiatric and neurological diseases and mental health; and a grant for nervous and mental disorders from the Ministry of Health, Labour and Welfare.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhang, Z., Habara, Y., Nishiyama, A. et al. Identification of seven novel cryptic exons embedded in the dystrophin gene and characterization of 14 cryptic dystrophin exons. J Hum Genet 52, 607–617 (2007). https://doi.org/10.1007/s10038-007-0163-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-007-0163-0
Keywords
This article is cited by
-
DMD transcripts in CRL-2061 rhabdomyosarcoma cells show high levels of intron retention by intron-specific PCR amplification
Cancer Cell International (2017)
-
Cryptic splice activation but not exon skipping is observed in minigene assays of dystrophin c.9361+1G>A mutation identified by NGS
Journal of Human Genetics (2017)
-
Targeted RNA-Seq profiling of splicing pattern in the DMD gene: exons are mostly constitutively spliced in human skeletal muscle
Scientific Reports (2017)
-
Normal and altered pre-mRNA processing in the DMD gene
Human Genetics (2017)
-
HEK293 cells express dystrophin Dp71 with nucleus-specific localization of Dp71ab
Histochemistry and Cell Biology (2016)
