
Abstract
The ability of glucocorticoids (GC) to efficiently kill lymphoid cells has led to their inclusion in essentially all chemotherapy procedures used to treat acute lymphoblastic leukemia (ALL). GC sensitivity is an important prognostic factor in ALL treatment, and it is used to classify patients into risk groups. Clinical assessment for GC sensitivity is very time-consuming, however. We have recently found that granzyme A (GZMA) mediates GC-induced apoptosis in ALL-derived cell line 697. In this study we examined the correlation between GC sensitivity and GC-induced GZMA expression by using seven established cell lines derived from ALL patients. The apoptosis assay showed four cell lines were GC-sensitive and three were GC-resistant. GC treatment markedly enhanced GZMA expression in GC-sensitive cell lines only, and not in GC-resistant cell lines. GC-induced GZMA expression also correlated well with the amount of GC-induced apoptosis. GC-induced GZMA expression could thus be a useful early biomarker for “personalized” ALL therapy.
Similar content being viewed by others


Introduction
Glucocorticoids (GC) are commonly used as chemotherapeutic agents to treat acute lymphoblastic leukemia (ALL) (Pui et al. 2004; Schmidt et al. 2004; Pui and Evans 2005). GC sensitivity is also used to classify patients for risk group-dependent therapy of ALL, because recent clinical studies have suggested that resistance to GC is associated with an unfavorable outcome in ALL (Dordelmann et al. 1999; Den Boer et al. 2003; Igarashi et al. 2005). The GC sensitivity of ALL is currently determined by monitoring blast cell reduction in peripheral blood after administration of GC for seven days (Igarashi et al. 2005). Clearly, it will be critical to detect the GC sensitivity earlier in the treatment to develop better and more “personalized” ALL treatment.
We recently examined changes in gene expression during dexamethasone (DEX)-induced apoptosis of GC-sensitive human pre-B ALL cell line 697 and found that granzyme A (GZMA) is critically involved in mediating the apoptotic effect of GC on leukemia cells (Yamada et al. 2003). GZMA has recently been shown to be a direct target gene of the glucocorticoid receptor (U et al. 2004; Ruike et al. 2006). The GZMA protein accumulated and its enzymatic activity increased in a time-dependent manner after DEX treatment, in accordance with the increase in caspase-3 activity (Yamada et al. 2003). To examine whether GC-induced GZMA expression could be an early biomarker for GC sensitivity, we investigated the relationship between GC-induced apoptosis and GZMA expression by using cell lines established from pre-B ALL patients in this study. The results showed that GC-induced GZMA expression at 24 h correlated well with GC sensitivity.
Materials and methods
Drugs
DEX and prednisolone (PRD) were purchased from Sigma Aldrich (St Louis, MO, USA). DEX and PRD were dissolved in dimethyl sulfoxide (DMSO; Nakalai Tesque, Kyoto, Japan) and 100% ethanol (Nakalai Tesque), respectively, and added to the medium. Each final concentration of DMSO and ethanol was adjusted to 0.1% (v/v). The control medium contained the same amount of DMSO or ethanol.
Cell lines
We analyzed seven established cell lines derived from pre-B ALL patients, in addition to 697 and 697Bcl2 as positive and negative controls, respectively. 697Bcl2 cells are 697 cells stably infected with a retrovirus encoding the anti-apoptotic protein Bcl-2 (Yamada et al. 2003; Miyashita and Reed 1993). It has previously been confirmed that over-expression of the Bcl-2 protein in the 697Bcl2 cells rendered leukemia cells highly resistant to apoptotic cell death induced by dexamethasone, although levels of granzyme A mRNA and protein in the cells were increased, showing that the granzyme A signaling induced by glucocorticoid acts upstream of Bcl-2 signaling in the apoptotic signaling pathway of glucocorticoid-treated leukemia cells. We therefore used 697Bcl2 cells as a negative control for glucocorticoid-induced apoptosis in leukemia 697 cells. All cell lines examined harbored a chromosomal translocation; thus, BV173 (Pegoraro et al. 1983), KOPN72bi, KOPN66bi, KOPN57bi (Inukai et al. 2000), and OM9;22 (Ohyashiki et al. 1993) harbor the t(9;22) translocation (known as BCR-ABL), the t(11;19) translocation (known as MLL-ENL) is harbored by KOCL33, and the t(4;11) translocation (known as MLL-AF4) is harbored by KOCL69 (Yamamoto et al. 1994). All cell lines were grown in RPMI-1640 medium supplemented with 10% heat-inactivated fetal calf serum, 50 U mL−1 penicillin, and 0.1 mg mL−1 streptomycin.
Cell-viability assays
All cells, growing at a cell density of 5 × 105 cells mL−1, were treated with DEX or PRD for 24 h. DEX was added to the media at different concentrations (0, 50, 100, 200 nmol L−1) and PRD was also added at different concentrations (0, 400, 800, 1,600 nmol L−1, respectively). Cell viability was assessed with the reagent WST-1, which involved measuring absorbance at 450 nm in a colorimetric assay using (4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate) (Roche Diagnostics GmbH, Mannheim, Germany) in accordance with the manufacturer’s procedure. All assays were independently performed six times and readings were averaged and plotted against drug concentration. All results are given as percentage mean viability for each pretreatment dose, and are summarized as mean ± SEM for each group.
Apoptosis assessment
All cells, growing at a cell density of 5 × 105 cells mL−1, were treated with or without 100 nmol L−1 DEX or 400 nmol L−1 PRD for 24 h. Cell death was defined at 0 or 24 h by propidium iodide (PI) staining; mitochondrial transmembrane potential (Δψ m) depolarization was determined by 3,3′-diexyloxacarbocyanine (DiOC6) staining as described previously (Okada et al. 2004). For each condition, 1.0 × 105 cells mL−1 were incubated with 5 mg mL−1 PI (Sigma Aldrich) and 20 nmol L−1 DiOC6 (Sigma Aldrich) at 37°C for 15 min, and their fluorescence was then measured using the FL1 and FL3 channels of a FACSCalibur (Becton Dickinson, Franklin Lakes, NJ, USA). All assays were independently performed four times. All measurements were normalized by mean viability of pretreatment, and were summarized as mean ± SEM for each group.
qRT-PCR
All cells, growing at a cell density of 5 × 105 cells mL−1, were treated with or without 100 nmol L−1 DEX or 400 nmol L−1 PRD for 24 h. At 0 or 24 h total RNA isolated from each sample was reverse-transcribed using the Quantitect reverse transcription kit (Qiagen, Valencia, CA, USA) and this was analyzed by quantitative PCR using ABsolute QPCR SYBR Green Mixes (ABgene, Surrey, UK) in a DNA Engine Opticon2 System (Biorad, Hercules, CA, USA) as described previously (Yamada et al. 2005). GAPDH was used to standardize the mRNA levels of target genes. Sequences of PCR primers were: for human GZMA (153 bp) the forward primer was 5′-caccatcatagacagaaaag-3′ and the reverse primer was 5′-gaaaacaccctcgcacaaca-3′; for human GAPDH (430 bp) the forward primer was 5′-tcaagatcatcagcaatgcc-3′ and the reverse primer was 5′-ttgaagtcagaggagaccac-3′. All assays were performed four times. All measurements were normalized by mean viability of pretreatment, and were summarized as mean ± SEM for each group.
Correlation between GZMA expression and apoptosis
The correlation between GC-induced GZMA expression and GC-induced apoptosis was assessed for each GC-sensitive cell line (Holleman et al. 2003). Briefly, each value was calculated using the formula: percentage of GZMA-positive or apoptotic cells = 100% × (C−D)/(100−D), where C represents the percentage of GZMA-positive or apoptotic cells in the presence of GCs and D is the percentage of GZMA-positive or apoptotic cells in the absence of GCs (spontaneous GZMA expression or apoptosis). All measurements were normalized by mean viability of pretreatment, and were summarized as mean ± SEM for each group.
Data analysis
Statistical analysis of the data was performed by using Dunnett’s test for multiple comparison and the two-tailed paired Student’s t-test. A P value <0.05 was regarded as statistically significant. Correlations between apoptosis assessment and qRT-PCR were calculated by using Pearson’s correlation and estimated from the linear regression line for all available data points. These analyses were performed using R, which is available at the Comprehensive R Archive Network site (http://www.cran.r-project.org).
Results and discussion
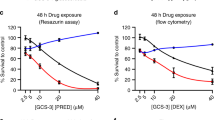
We first assessed the GC sensitivity of seven established cell lines derived from ALL patients. 697 leukemia cells were treated with or without different concentrations of GCs for several periods of time and the viability of the cells was determined by WST-1 assay. After 24 h there was a significant (P < 0.01) decrease in the number of viable cells treated with at least 50 nmol L−1 DEX or 400 nmol L−1 PRD. These decreases were even more pronounced 48 h after treatment whereas the untreated cells were viable and growth was normal during the 48 h (Figs. 1A, B). It is suggested DEX is five to six times more cytotoxic than PRD in the anti-inflammatory activity of these drugs (Ito et al. 1996). These results indicated that DEX and PRD effectively induced cell death in 697 cells at concentrations of at least 50 and 400 nmol L−1, respectively. Moreover, 100 nmol L−1 DEX and 800 nmol L−1 PRD are comparable with clinically therapeutic levels (Miyashita and Reed 1993; Turner et al. 1973). For this study we therefore used DEX and PRD at 100 and 800 nmol L−1, respectively.

Induction of cell death in 697 cells treated with DEX and PRD. 697 leukemia cells were treated with or without different concentrations of DEX (A) and PRD (B) for several periods of time and cell viability was then determined by use of the WST-1 assay. To compare independent experiments, all data are given as percentage mean viability of pretreatment cells. (A) Open circles 0 nmol L−1 DEX, filled circles 50 nmol L−1 DEX, filled triangles 100 nmol L−1 DEX, multiplication symbols 200 nmol L−1 DEX. (B) Open circles 0 nmol L−1 PRD, filled circles 400 nmol L−1 PRD, filled triangles 800 nmol L−1 PRD, multiplication symbols 1,600 nmol L−1 PRD. The mean and SEM for six independent experiments are shown. **P < 0.01 compared with untreated cells (Dunnett’s test)
As shown in Fig. 2A, GC treatment significantly (P < 0.05) enhanced cell death in BV173, KOPN72bi, KOPN66bi, and OM9;22, but not in KOPN57bi, KOCL33, or KOCL69. The same GC treatment also significantly (P < 0.01) enhanced Δψ m depolarization in BV173, KOPN72bi, KOPN66bi, and OM9;22, but not in KOPN57bi, KOCL33, or KOCL69 (Fig. 2B). In all experiments, DEX treatment markedly enhanced both cell death and Δψ m depolarization for 697 cells whereas it had no effect on 697Bcl2 cells (Figs. 2A, B). We also examined the effect of PRD on cell death and Δψ m depolarization, because it is frequently used with DEX for GC therapy to treat ALL. Similar to DEX, PRD significantly (P < 0.01) enhanced both cell death and Δψ m depolarization in BV173, but not in KOPN57bi. Because enhanced cell death and Δψ m depolarization responses are regarded as apoptotic, these results may indicate that cell lines BV173, KOPN72bi, KOPN66bi, and OM9;22 are GC-sensitive whereas cell lines KOPN57bi, KOCL33, and KOCL69 are GC-resistant.

GC sensitivity among seven established cell lines derived from ALL patients. All cell lines were treated for 24 h with GC (left 100 nmol L−1 DEX and right 800 nmol L−1 PRD; black bars) or vehicle (white bars). Cell death and Δψ m depolarization were detected by PI-staining (A) and DiOC6-staining (B), respectively. Differences between pretreatment values and DEX or vehicle treatment values are shown as mean ± SEM of results from four independent experiments. *P < 0.05; **P < 0.01 compared with vehicle (Student’s t-test). Before treatment the percentage cell death and the percentage Δψ m depolarization were 2.3% ± 0.8 and 7.5% ± 1.1 for BV173, 1.5% ± 0.2 and 6.5% ± 0.9 for KOPN72bi, 1.0% ± 0.1 and 3.7% ± 0.1 for KOPN66bi, 3.8% ± 0.9 and 6.13% ± 1.9 for OM9; 22, 1.2% ± 0.3 and 6.1% ± 1.4 for KOPN57bi, 5.0% ± 0.9 and 12.6% ± 2.4 for KOCL33, and 1.7% ± 0.4 and 6.5% ± 1.6 for KOCL69, all respectively
We next examined the effect of GC treatment on GZMA mRNA expression in these seven cell lines using qRT-PCR. As shown in Fig. 3, DEX treatment significantly (P < 0.05) enhanced GZMA expression in BV173, KOPN72bi, KOPN66bi, and OM9;22, although the amount of enhanced GZMA expression differed, depending on cell line. DEX had no effect on GZMA expression in the KOPN57bi, KOCL33, or KOCL69 cell lines, however. In all experiments DEX treatment significantly enhanced GZMA expression in both 697 and 697Bcl2 cells (Fig. 3), confirming that GZMA signaling induced by GC acts upstream of BCL-2 signaling in the apoptotic signaling pathway of GC-treated 697 cells (Yamada et al. 2003). We also found that PRD significantly (P < 0.01) enhanced GZMA expression in the BV173 cell line but not in the KOPN57bi cell line. We examined the correlations between GC sensitivity and GC-induced GZMA expression. As summarized in Fig. 4, highly significant correlation was observed between GC-induced GZMA expression and apoptosis, showing that the 24-h GC-induced GZMA expression was correlated with the GC-sensitivity of ALL cells.

GZMA mRNA expression in seven established cell lines derived from ALL patents. All cell lines were treated for 24 h with GC (left 100 nmol L−1 DEX and right 800 nmol L−1 PRD, black bars) or vehicle (white bars). GZMA mRNA expression was measured by qRT-PCR. The GAPDH gene was used as a control. Differences between the pretreatment values and DEX or vehicle treatment values are shown as mean ± SEM of results from four independent experiments. *P < 0.05; **P < 0.01 compared with vehicle (Student’s t-test). Before treatment, percentage GZMA expression was 0.0052 ± 0.001% for BV173, 0.0001 ± 0.003% for KOPN72bi, 0.3910 ± 0.003% for KOPN 66bi, 0.0020 ± 0.002% for OM9;22, 0.0020 ± 0.001% for KOPN57bi, 0.0880 ± 0.017% for KOCL33, and 0.0140 ± 0.005% for KOCL69

Correlation between GC-induced apoptosis and GC-induced GZMA mRNA expression. Correlation between GZMA mRNA expression and GC-induced cell death (A) and GC-induced Δψ m depolarization (B). Each value was calculated using the formula: 100% × (C−D)/(100−D), where C and D represent the percentage cell death, Δψ m depolarization, or GZMA expression in GC-treated and vehicle-treated samples, respectively. Percentages compared with vehicle treatment are shown as mean ± SEM of results from three independent experiments. The solid line represents the linear regression line
In this study we found a highly significant correlation between GC-induced GZMA expression and apoptosis at 24 h by using seven cell lines established from ALL patients. In a previous study we examined the protein granzyme A, and found activation was induced by glucocorticoid treatment in 697 and 697Bcl2. As described in the Introduction, GCs are essential in the treatment of ALL, and GC sensitivity is now used to classify patients receiving risk group-dependent therapy to treat ALL (Pui et al. 2004; Schmidt et al. 2004; Pui and Evans 2005); the current method for assessing GC sensitivity requires a long period of time (more than 1 week; Dordelmann et al. 1999; Den Boer et al. 2003; Igarashi et al. 2005), however. Clearly, early detection of GC-sensitivity/GC-resistance would be of potential benefit in selecting the most appropriate drug regimens for ALL patients.
In our opinion GC-induced GZMA expression could be a potentially important candidate for such a biomarker. To establish GC-induced GZMA expression as an early biomarker of glucocorticoid sensitivity of ALL therapy, however, some problems must still be resolved. This study was conducted on cultured cell lines only, so primary ALL samples and prospective and/or retrospective studies should be performed from the viewpoint of human genetics. Technical improvements for rapid and accurate monitoring of GZMA expression are also urgently required for clinical use. Further studies will clearly be required to establish GC-induced GZMA expression as an early biomarker for “personalized” ALL therapy.
Abbreviations
- ALL:
-
Acute lymphoblastic leukemia
- GZMA:
-
Granzyme A
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- DEX:
-
Dexamethasone
- PRD:
-
Prednisolone
- PI:
-
Propidium iodide
- DiOC6 :
-
3,3′-Diexyloxacarbocyanine
- DMSO:
-
Dimethyl sulfoxide
- qRT-PCR:
-
Quantitative reverse transcriptase-polymerase chain reaction
References
Den Boer ML, Harms DO, Pieters R, Kazemier KM, Gobel U, Korholz D, Graubner U, Haas RJ, Jorch N, Spaar HJ, Kaspers GJ, Kamps WA, Van der Does-Van den Berg A, Van Wering ER, Veerman AJ, Janka-Schaub GE (2003) Patient stratification based on prednisolone-vincristine-asparaginase resistance profiles in children with acute lymphoblastic leukemia. J Clin Oncol 21:3262–3268
Dordelmann M, Reiter A, Borkhardt A, Ludwig WD, Gotz N, Viehmann S, Gadner H, Riehm H, Schrappe M (1999) Prednisone response is the strongest predictor of treatment outcome in infant acute lymphoblastic leukemia. Blood 94:1209–1217
Holleman A, den Boer ML, Kazemier KM, Janka-Schaub GE, Pieters R (2003) Resistance to different classes of drugs is associated with impaired apoptosis in childhood acute lymphoblastic leukemia. Blood 102:4541–4546
Igarashi S, Manabe A, Ohara K, Kumagai M, Saito T, Okimoto Y, Kamijo T, Isoyama K, Kajiwara M, Sotomatsu M, Sugita K, Sugita K, Maeda M, Yabe H, Kinoshita A, Kaneko T, Hayashi Y, Ikuta K, Hanada R, Tsuchida M (2005) No advantage of dexamethasone over prednisolone for the outcome of standard- and intermediate-risk childhood acute lymphoblastic leukemia in the Tokyo Children’s Cancer Study Group L95-14 protocol. J Clin Oncol 23:6489–6498
Inukai T, Sugita K, Mitsui K, Iijima K, Goi K, Tezuka T, Kojika S, Kagami K, Mori T, Kinoshita A, Suzuki T, Okazaki-Koyama T, Nakazawa S (2000) Participation of granulocyte colony-stimulating factor in the growth regulation of leukemia cells from Philadelphia chromosome-positive acute leukemia and blast crisis of chronic myeloid leukemia. Leukemia 14:1386–1395
Ito C, Evans WE, McNinch L, Coustan-Smith E, Mahmoud H, Pui CH, Campana DJ (1996) Comparative cytotoxicity of dexamethasone and prednisolone in childhood acute lymphoblastic leukemia. Clin Oncol 8:2370–6
Miyashita T, Reed JC (1993) Bcl-2 oncoprotein blocks chemotherapy-induced apoptosis in a human leukemia cell line. Blood 81:151–157
Ohyashiki K, Miyauchi J, Ohyashiki JH, Saito M, Yaguchi M, Inatomi Y, Nakazawa S, Waca H, Mizutani S, Matsuo Y (1993) Interleukin-7 enhances colony growth and induces CD20 antigen of a Ph+ acute lymphoblastic leukemia cell line, OM9;22. Leukemia 7:1034–1040
Okada M, Adachi S, Imai T, Watanabe K, Toyokuni SY, Ueno M, Zervos AS, Kroemer G, Nakahata T (2004) A novel mechanism for imatinib mesylate-induced cell death of BCR-ABL-positive human leukemic cells: caspase-independent, necrosis-like programmed cell death mediated by serine protease activity. Blood 103:2299–2307
Pegoraro L, Matera L, Ritz J, Levis A, Palumbo A, Biagini G (1983) Establishment of a Ph1-positive human cell line (BV173). J Natl Cancer Inst 70:447–453
Pui CH, Evans WE (2005) Treatment of acute lymphoblastic leukemia. N Engl J Med 354:166–178
Pui CH, Relling MV, Downing JR (2004) Acute lymphoblastic leukemia. N Engl J Med 350:1535–1548
Ruike Y, Katsuma S, Hirasawa A, Tsujimoto G (2006) Glucocorticoid-induced alternative promoter usage for a novel 5′ variant of granzyme A. J Hum Genet. December 19 2006; (Epub ahead of print)
Schmidt S, Rainer J, Ploner C, Presul E, Riml S, Kofler R (2004) Glucocorticoid-induced apoptosis and glucocorticoid resistance: molecular mechanisms and clinical relevance. Cell Death Differ 11(Suppl 1):S45–S55
Turner AK, Carroll CJ, Pinkus JL, Charles D, Chattoraj SC (1973) Simultaneous competitive protein binding assay for cortisol, cortisone, and prednisolone in plasma, and its clinical application. Clin Chem 19:731–736
U M, Shen L, Oshida T, Miyauchi J, Yamada M, Miyashita T (2004) Identification of novel direct transcriptional targets of glucocorticoid receptor. Leukemia 18:1850–1856
Yamada M, Hirasawa A, Shiojima S, Tsujimoto G (2003) Granzyme A mediates glucocorticoid-induced apoptosis in leukemia cells. FASEB J 17:1712–1714
Yamada M, Katsuma S, Adachi T, Hirasawa A, Shiojima S, Kadowaki T, Okuno Y, Koshimizu TA, Fujii S, Sekiya Y, Miyamoto Y, Tamura M, Yumura W, Nihei H, Kobayashi M, Tsujimoto G (2005) Inhibition of protein kinase CK2 prevents the progression of glomerulonephritis. Proc Natl Acad Sci USA 102:7736–7741
Yamamoto K, Seto M, Iida S, Komatsu H, Kamada N, Kojima S, Kodera Y, Nakazawa S, Saito H, Takahashi T, Ueda R (1994) A reverse transcriptase-polymerase chain reaction detects heterogeneous chimeric mRNAs in leukemias with 11q23 abnormalities. Blood 83:2912–2921
Acknowledgments
This work was supported in part by research grants from the Scientific Fund of the Ministry of Education, Science, and Culture of Japan (to G.T.and T.K.); the Ministry of Human Health and Welfare (to G.T. and T.K.); and the 21st Century Center of Excellence Program “Knowledge Information Infrastructure for Genome Science” (to G.T.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Myoumoto, A., Nakatani, K., Koshimizu, Ta. et al. Glucocorticoid-induced granzyme A expression can be used as a marker of glucocorticoid sensitivity for acute lymphoblastic leukemia therapy. J Hum Genet 52, 328–333 (2007). https://doi.org/10.1007/s10038-007-0119-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-007-0119-4
Keywords
This article is cited by
-
SGAClust: Semi-supervised Graph Attraction Clustering of gene expression data
Network Modeling Analysis in Health Informatics and Bioinformatics (2022)
