
Abstract
Propionic acidemia results from mutations in either of the two genes, PCCA or PCCB, that encode the two subunits of the propionyl-CoA carboxylase (PCC) enzyme. In this study, we report the identification and analysis of seven novel splicing mutations involving consensus donor and acceptor splice sites. Most of them were identified in patients with a Central Asian origin, and some present in several alleles, probably reflecting founder effects. The functional consequences of the splicing mutations were analyzed in patients’ fibroblasts, as well as transcript quantification using real-time PCR methods. In the PCCA gene, two mutations were demonstrated to affect 5′ splice sites (c.231+1G>C and c.1209+3A>G) and two 3′ acceptor splice sites (c.1210delG and c.1430G>T), all causing skipping of the exons involved, with no detectable levels of normally spliced transcript. In the PCCB gene, all three mutations involved 5′ donor splice sites—two affected exon 1 splicing (c.154_183+17del46 and c.183+2T>C), the latter activating a cryptic splice site in intron 1, and the remaining mutation (c.1498+2T>C) resulted in exon 14 skipping. The results highlight the necessity to perform transcript analysis in addition to genomic DNA sequencing to characterize the effect of splicing mutations and add relevant information on the genetic epidemiology of the disease.
Similar content being viewed by others


Introduction
Propionic acidemia (PA: OMIM#606054) is one of the most frequent organic acidemias caused by a defect of propionyl-CoA carboxylase (PCC, E.C.6.4.1.3). It is inherited in an autosomal recessive fashion and caused by mutations in either of the two genes, PCCA (OMIM 232000) or PCCB (OMIM 232050), coding for the α and β subunits, respectively, of the PCC enzyme. The disease can be fatal with a neonatal presentation, although other patients show a later onset with a better outcome (Fenton et al. 2001). PA is highly heterogeneous at the genetic level, especially for Caucasian PCCA-deficient patients with no prevalent mutations, while for the PCCB gene a limited number of mutations accounts for most of the mutant alleles. A mutation update and review of the expression analysis of PCCA and PCCB mutations has been published recently (Desviat et al. 2004). In both genes, missense mutations are the most frequent defects (approximately 40%) followed by small insertions/deletions and splicing mutations, most of which result in frameshift. The mutational spectrum is clearly population specific. Several Caucasian and Oriental populations have been genetically analyzed, and some mutations have been found to be exclusively associated with geographical or ethnic groups (PCCB mutations E168K in Spanish chromosomes, A513_R514insP in the Inuit population, T428I in Orientals, etc)(Desviat et al. 2004).
According to the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php), approximately 10% of the disease-causing mutations in humans affects consensus splice sites (3′ acceptor, 5′ donor sites and branch site). In addition, mutations affecting auxiliary splicing sequences within exons or introns (exonic or intronic splicing enhancers or silencers, ESE, ESS, ISE and ISS) are also being increasingly identified as splicing mutations (Baralle and Baralle 2005; Cartegni et al. 2002). Splicing mutations can lead to skipping of the involved exon or to activation of cryptic splice sites, and sometimes to a mixture of aberrantly and correctly spliced transcripts; so, without transcript analysis in patients’ cells or in adequate splicing cellular models (minigenes), it is impossible to predict the consequences of a given mutation potentially affecting splicing. Relative to protein function, most splicing aberrations usually result in disruption of the open reading frame, introducing a premature termination codon (PTC), thus destabilizing mRNA through nonsense-mediated decay (NMD)(Maquat 2004). The level of correctly spliced transcript and the ratio of splicing variants has been described to vary in different tissues and among individuals, correlating with disease severity (Nissim-Rafinia and Kerem 2002).
To gain insight into the functional consequences of splicing mutations and their relationship to the phenotype, in this study, we analyzed seven PA mutations not previously reported and affecting consensus splice sites. Most mutations were found in homozygous fashion and some are present in several unrelated patients, probably reflecting consanguinity and a common origin related to the ethnic or geographical background of the patients. The effect of the mutations on mRNA splicing and the different transcript levels was analyzed in patients’ fibroblasts.
Patients and methods
This study included ten previously diagnosed PA patients referred from different countries for genetic diagnosis. Samples used as source of DNA and/or RNA included whole blood, dried blood spots and/or fibroblast cell lines. When fibroblast cell lines were available, transfection studies using PCCA or PCCB cDNAs were performed (Pérez-Cerdá et al. 2002) to identify the defective gene, PCCA or PCCB, prior to genetic analysis. Subsequently, mutations were detected by direct sequencing of amplified fragments from genomic DNA and/or cDNA of the corresponding gene. Intronic and exonic primers for amplification of cDNA and genomic sequences of both genes have been described previously (Campeau et al. 2001; Richard et al. 1999; Rodriguez-Pombo et al. 1998). The PCR products were sequenced with BigDye Terminator v.3.1 mix (Applied BioSystems), and products were analyzed by means of capillary electrophoresis on an ABI Prism 3700 Genetic Analyzer (Applied Biosystems). DNA was sequenced with the same primers used for amplification.
When parental samples were available, Mendelian inheritance was confirmed by direct sequencing of amplified products from the corresponding region. In these cases, the parental analysis also confirmed that the mutations were present in homozygous fashion.
For quantification of normal transcripts, total RNA was isolated from patients and control fibroblasts using the Tripure reagent (Roche). One microgram of RNA was retrotranscribed using Superscript III First-Strand Synthesis System (Life technologies) and subsequently amplified by means of real-time PCR, using SYBR Green and the LightCycler instrument (Roche Diagnostics). PCR was performed in a total volume of 20 μl, containing 2–8 μl cDNA product, 2 μl LightCycler FastStart DNA Master SYBR Green I, 1.6 μl MgCl2 25 mM and 0.5 μM of each primer. To selectively amplify normal transcripts without exon skipping, exon specific primers were used in each case (available on request). The amplification parameters were as follows: denaturation at 95°C for 10 min, followed by 45 cycles of amplification (95°C for 10 s, 55–60°C for 5 s, 72°C for 10 s, 82–86°C for 2 s, single acquisition mode) and termination by melting (95°C for 0 s, 65°C for 15 s and 95°C for 0 s). The data were analyzed by the second derivative method, using the LightCycler software, to correlate relative initial template concentration to the crossing point (Cp), which is the cycle number where the exponential amplification begins. In other cases, the quantitative analysis was performed by means of retrotranscription and real-time PCR using Taqman probe Hs01120554_m1, specific for PCCA exon 13 sequences, supplied by Applied Biosystems (Taqman® Gene Expression Assays, http://www.myscience.appliedbiosystems.com). The retrotranscription was performed using 10 ng total RNA/μl and the Archive kit from Applied Biosystems. The TaqMan® 2× Universal PCR Master Mix No AmpErase UNG, also provided by Applied Biosystems, was used for amplification, following the instructions of the manufacturer. The relative efficiency of amplification was monitored by performing the analysis in parallel with GAPDH mRNA or 18S rRNA. The real-time PCR and analysis were performed in an ABIPRISM 7900HT Genetic Analyzer (Applied Biosystems) using the SDS2.1 program. The analysis determines the relative quantity (RQ), defined as RQ=2−ΔΔCt , where ΔΔCt=ΔCtsample−ΔCtcontrol and ΔCt=Ctsample−CtGAPDH/18SrRNA. Ct is defined as the “cycling threshold”, analogous to Cp.
Results and discussion
In this study, we report seven novel PA mutations, four in the PCCA gene and three in the PCCB gene (Table 1), all corresponding to point mutations or small deletions involving consensus splice sites and causing different splicing defects, presumably pathogenic. The mutations were identified in a cohort of ten patients, six PCCA deficient and four PCCB deficient. Many patients (6 of 10) were homozygous, which could be due to consanguinity between the parents, although this information was not available. Two mutations (c.1209+3A>G in the PCCA gene and c.1498+2T>C in the PCCB gene) were detected in more than one patient with a common geographical or ethnic origin, probably reflecting founder effects (Table 1).
In the PCCA gene, the c.231+1G>C changes the invariant 5′ donor site of exon 3, lowering the Shapiro score (Shapiro and Senapathy 1987) from 88.9 to 70.6. Intron 3 is small, 104 bp, and the splice sites of exon 4 are weak, probably determining that both exons 3 and 4 are skipped together as a consequence of the mutation. Skipping of both exons in cDNA has also been observed in other patients, as in 19218 included in this study, with a yet unidentified causative mutation in genomic DNA (Desviat et al. 2004).
The novel PCCA mutation c.1209+3A>G is a transition affecting the 5′ donor splice site of exon 13. At the cDNA level, the skipping of exon 13 is observed, which predictably originates a protein with an internal deletion of 48 amino acids. This change has been detected in two patients of Turkish origin (one homozygous and one heterozygous) and in one homozygous Afghan patient, the latter being up to date asymptomatic after being detected in a neonatal tandem mass screening program. The Shapiro score for the normal donor splice site of exon 13 is 79.4, whereas it decreases to 76.1 with the mutation. This change is not too drastic, as both A and G are similarly conserved at the +3 position according to Shapiro and Senapathy (1987). Taking this into account, we investigated the possibility of the presence of some normal transcript in patients’ fibroblasts. For that purpose, we isolated RNA from fibroblasts of patients homozygous for this change and quantified the presence of exon 13-containing (i.e., normal) transcripts by means of real-time PCR using two methodologies: (1) amplification using an oligonucleotide hybridizing to exon 13 and the double helix-specific dye SYBR Green I; and (2) amplification and detection with a Taqman probe specific for the cDNA region corresponding to the exon 13–14 junction. Although both approaches are rapid and sensitive, their underlying principles are different and, in some cases, SYBR Green I has been reported to provide reliable quantification only when the target gene is expressed at moderate to high levels, in contrast to Taqman probes (Yin et al. 2001). Other studies have reported similar sensitivity for both methods (Hembruff et al. 2005). Using the SYBR Green I methodology, we were not able not detect any normal transcript from the c.1209+3A>G allele when comparing with control samples. The amplification was not successful enough, as several unspecific products were amplified in the patients’ samples. Using a Taqman probe specific for exons 13–14 containing sequences, we could detect extremely low levels of normal transcripts in fibroblasts from the two homozygous patients. The relative quantity of normal mRNAs relative to that in controls was quantified as 1.8% and 2.7% for the two homozygous patients (Fig. 1). Madsen et al. (2006) recently pointed out that a +3A>G mutation causes exon skipping only in the context of a suboptimal 5′ splice site, with non-conserved bases mainly at positions +4 and +5, in order to ensure efficient base pairing to U1 snRNA during the splicing process. In agreement with this, the 5′ splice site of PCCA exon 13 has non-matching nucleotides to U1 snRNA at positions +5 and +6 (Fig. 1), which may account for its dependence on the presence of an A at the +3 position. Given that the amount of normal transcript is negligible in the asymptomatic patient homozygous for this mutation, other factors, such as increased flow through secondary pathways preventing PA accumulation (Fenton et al. 2001) or resulting protein with partial activity, may explain the milder course of the disease. Interindividual or intertissue variation in splicing factors can also be responsible for phenotypic variation (Nissim-Rafinia and Kerem 2002). Indeed, one has to bear in mind the limitation in studying fibroblasts as the only available sample, given that different levels of transcripts have been described in different tissues of the same individual (Nissim-Rafinia et al. 2000).

Real-time PCR quantification of normally spliced transcripts derived from the c.1209+3A>G PCCA mutation and effect on the binding of the U1 snRNA to the 5′ splice site of exon 13. a Relative quantification plot in a logarithmic scale (logRQ). RQ=2−ΔΔCT. ΔΔCt is calculated as difference between ΔCt of target and ΔCt of internal control gene (GAPDH). The quantities are shown relative to the expression level in a control sample. The data are the mean from three different experiments. b The pairing of wild type and mutant (c.1209+3A>G) 5′ splice site of exon 13 of the PCCA gene with the U1 snRNA is shown. A vertical line shows complementary base pairing and semicolon indicates mismatched bases
The PCCA change c.1210delG is a deletion of a guanidine in the first base of exon 14, included in the 3′ acceptor splice site. It was detected in a homozygous fashion in a Canadian patient of Pakistani origin. This deletion lowers the Shapiro splicing score from 91.5 to 88. At the cDNA level, we could observe the skipping of both exons 13 and 14. This predictably originates a protein with an internal deletion of 73 amino acids.
The c.1430G>T mutation changes the first base of exon 17 in the PCCA gene, thus lowering the 3′ splice acceptor site score from 88.7 to 82.4. This change causes skipping of exon 17. The patient is heterozygous with a missense mutation on the other allele, therefore the possibility of normal transcript resulting from the splicing mutation could not be assayed.
In the PCCB gene, we identified a 46-bp deletion encompassing the last 29 nucleotides of exon 1 plus the first 17 intronic bases (c.154_183+17del46, Fig. 2). Only DNA samples were available from the patient, thus precluding transcript analysis, although the mutation can be assumed to disrupt exon 1 splicing due to the abolition of the 5′ splice site. Examination of the sequences surrounding the deletion shows the presence of a direct repeat sequence GGGGCC (Fig. 2), suggesting that the deletion occurred through a slipped strand mispairing mechanism, as in other deletions causing human disease (Chuzhanova et al. 2003; Darvasi and Kerem 1995).

Sequencing analysis of patient heterozygous for the deletion c.154_183+17del46 in the PCCB gene. The deletion includes nucleotides 154–183 of the coding sequence (exon 1) plus the first 17 nucleotides of intron 1. Direct repeat sequences at both sides of the deletion are underlined
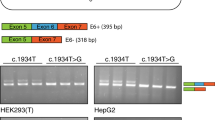
The c.183+2T>C also affected the 5′ donor splice site of exon 1, changing the invariant GT. This change caused the activation of a cryptic donor splice site located downstream in intron 1 (Fig. 3). The Shapiro scores for the wild-type, mutant and cryptic splice site are 76.5, 58.2 and 84.3, respectively. The use of the cryptic splice site caused the insertion of 20 bp of intron 1, which would originate a frameshift and an aberrant PCCB protein. This aberrant mRNA was the only one we could rescue using an oligonucleotide specific for exon 1 and real-time PCR methodology in the LightCycler instrument. The mRNA with the intronic insertion was present in patients' fibroblasts in a proportion of 9–18% relative to the amount of normal transcripts found in a control (100%). This transcript predictably resulting in a prematurely truncated protein is probably subjected to NMD (Maquat 2004), which explains its low levels.

Effect of the PCCB mutation c.183+2T>C. The mutation affects the thymine at the invariant GT of the 5′ donor splice site of exon 1 of the PCCB gene. This causes the activation of a cryptic donor splice site located downstream and the inclusion of 20 bp from intron 1 at the cDNA level, which would cause a frameshift and an aberrant PCCB protein. The Shapiro scores for the wild-type, mutant and cryptic splice-sites are indicated between brackets
The other novel PCCB change c.1498+2T>C was detected in two patients whose families originated in Iran and Pakistan. This transition is affecting one of the invariant bases at the 5′ donor splice site of PCCB exon 14, which reduces the splicing score from 84.7 to 66.9 according to Shapiro and Senapathy (1987). In patients’ fibroblasts we could observe, at the cDNA level, the skipping of exon 14 of the PCCB gene, which would originate a frameshift. We investigated the possibility of the presence of some normal transcript in patients’ fibroblasts. For that purpose, we designed an oligonucleotide specific for exon 14, and RNA from patients’ fibroblasts was analyzed using RT- and real-time PCR with SYBR Green I. With this method, the amount of normal spliced transcripts in both patients’ fibroblasts was less than 0.1% of the proportion of normal transcripts obtained in a control (100%).
The results obtained in this study reflect the different consequences derived from splicing mutations, which, in turn, can be influenced by complex networks of splicing factors differentially expressed in each tissue and/or individual, thus resulting in phenotypic variability. Investigation of the functional consequences of splicing mutations is essential for the development of molecular therapies aimed at the correction of erroneous splicing.
References
Baralle D, Baralle M (2005) Splicing in action: assessing disease causing sequence changes. J Med Genet 42:737–748
Campeau E, Desviat LR, Leclerc D, Wu X, Perez B, Ugarte M, Gravel RA (2001) Structure of the PCCA gene and distribution of mutations causing propionic acidemia. Mol Genet Metab 74:238–247
Cartegni L, Chew SL, Krainer AR (2002) Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet 3:285–298
Chuzhanova N, Abeysinghe SS, Krawczak M, Cooper DN (2003) Translocation and gross deletion breakpoints in human inherited disease and cancer II: potential involvement of repetitive sequence elements in secondary structure formation between DNA ends. Hum Mutat 22:245–251
Darvasi A, Kerem B (1995) Deletion and insertion mutations in short tandem repeats in the coding regions of human genes. Eur J Hum Genet 3:14–20
Desviat LR, Perez B, Perez-Cerda C, Rodriguez-Pombo P, Clavero S, Ugarte M (2004) Propionic acidemia: mutation update and functional and structural effects of the variant alleles. Mol Genet Metab 83:28–37
Fenton WA, Gravel RA, Rosenberg LE (2001) Disorders of propionate and methylmalonate metabolism. In: Scriver CR, Beaudet AL, Sly W, Valle D (eds) The metabolic and molecular bases of inherited disease, 8th edn. McGraw-Hill, New York, pp 2165–2190
Hembruff SL, Villeneuve DJ, Parissenti AM (2005) The optimization of quantitative reverse transcription PCR for verification of cDNA microarray data. Anal Biochem 345:237–249
Madsen PP, Kibaek M, Roca X, Sachidanandam R, Krainer AR, Christensen E, Steiner RD, Gibson KM, Corydon TJ, Knudsen I, Wanders RJ, Ruiter JP, Gregersen N, Andresen BS (2006) Short/branched-chain acyl-CoA dehydrogenase deficiency due to an IVS3 + 3A > G mutation that causes exon skipping. Hum Genet 118(6):680–690
Maquat LE (2004) Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol 5:89–99
Nissim-Rafinia M, Chiba-Falek O, Sharon G, Boss A, Kerem B (2000) Cellular and viral splicing factors can modify the splicing pattern of CFTR transcripts carrying splicing mutations. Hum Mol Genet 9:1771–1778
Nissim-Rafinia M, Kerem B (2002) Splicing regulation as a potential genetic modifier. Trends Genet 18:123–127
Pérez-Cerdá C, Desviat LR, Perez B, Ugarte M, Rodriguez-Pombo P (2002) Transfection screening for defects in the PCCA and PCCB genes encoding propionyl-CoA carboxylase subunits. Mol Genet Metab 75:276–279
Richard E, Desviat LR, Perez B, Perez-Cerda C, Ugarte M (1999) Genetic heterogeneity in propionic acidemia patients with alpha-subunit defects. Identification of five novel mutations, one of them causing instability of the protein. Biochim Biophys Acta 1453:351–358
Rodriguez-Pombo P, Hoenicka J, Muro S, Perez B, Perez-Cerda C, Richard E, Desviat LR, Ugarte M (1998) Human propionyl-CoA carboxylase beta subunit gene: exon-intron definition and mutation spectrum in Spanish and Latin American propionic acidemia patients. Am J Hum Genet 63:360–369
Shapiro MB, Senapathy P (1987) RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res 15:7155–7174
Yin JL, Shackel NA, Zekry A, McGuinness PH, Richards C, Putten KV, McCaughan GW, Eris JM, Bishop GA (2001) Real-time reverse transcriptase-polymerase chain reaction (RT-PCR) for measurement of cytokine and growth factor mRNA expression with fluorogenic probes or SYBR Green I. Immunol Cell Biol 79:213–221
Acknowledgments
We thank the following: Drs Boneh (Melbourne), Haase (Jena), Hankinson (USA), Hewson (Ontario), Muschol and Mulhausen (Hamburg), Parlowsky (Lübeck), Poplawski (Ontario), Ribes (Barcelona) and Sewell (Frankfurt) for sending samples from their patients. The authors wish to thank C. Hernández, A. Sánchez and M.J. Ecay for expert technical assistance. This work was supported by grants from the Instituto de Salud Carlos III, Ministerio de Sanidad y Consumo (PI05/1403 and PI05/1413) and from the Comisión Interministerial de Ciencia y Tecnología (SAF2004-06298). The institutional grant from Fundación Ramón Areces to Centro de Biología Molecular is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Desviat, L.R., Clavero, S., Perez-Cerdá, C. et al. New splicing mutations in propionic acidemia. J Hum Genet 51, 992–997 (2006). https://doi.org/10.1007/s10038-006-0068-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-006-0068-3
Keywords
This article is cited by
-
Clinical characteristics and mutation analysis of propionic acidemia in Thailand
World Journal of Pediatrics (2014)
-
Functional characterization of novel genotypes and cellular oxidative stress studies in propionic acidemia
Journal of Inherited Metabolic Disease (2013)
-
Structure and function of biotin-dependent carboxylases
Cellular and Molecular Life Sciences (2013)
-
Crystal structure of the α6β6 holoenzyme of propionyl-coenzyme A carboxylase
Nature (2010)
-
Propionazidämie und Schallempfindungsschwerhörigkeit
HNO (2008)
