
Abstract
Mutations in the VHL gene cause von Hippel–Lindau disease, a cancer predisposing syndrome characterized by a variety of benign and malignant neoplasms. We report the molecular characterization of two sequence variants of the VHL gene: a synonymous substitution c.462 A>C in exon 2 and a duplication of 11 bp in the promoter region (c.−65_−55dup11). The first variant is a pathogenic mutation because, although it does not change the sense of the affected codon, it causes skipping of exon 2 in the affected allele by altering the splicing consensus site at the 3′ end of exon 2. The 11 bp duplication represents a nonpathogenic variant. In fact, although it affects a critical region of the VHL promoter, it was found in healthy controls, and we show that carrier individuals express both VHL alleles at equimolar levels. Our data underline the importance of careful evaluation of the potential pathogenicity of sequence variants that may not belong to the obvious disease-causing mutation categories, or that affect relevant regulatory regions. mRNA analysis will be required to ultimately resolve this issue.
Similar content being viewed by others

Introduction
The VHL gene (GenBank NM_000551) is a tumor suppressor gene located on chromosome 3p25 that encodes a 213 aa protein belonging to the ubiquitin ligase family. Mutations in VHL cause von Hippel–Lindau disease (MIM 193300), a dominantly inherited familial cancer syndrome predisposing to a variety of malignant and benign neoplasms, including retinal, cerebellar, and spinal hemangioblastoma, clear cell renal carcinoma and pheochromocytoma (Maher et al. 1991).
The disease is typically manifest after the second decade of life, although earlier onset is frequent. Penetrance is virtually complete by 60 years of age.
To date, missense, nonsense and frameshift mutations have been associated with the disease, as well as large-scale deletions and intronic mutations that alter mRNA splicing (Maher et al. 1991; Kaelin and Maher 1998); the pathogenicity of some sequence variants of the VHL gene is, however, still difficult to establish.
In this work we describe the molecular characterization of two such variants: a synonymous substitution in exon 2, and the duplication of 11 nucleotides in the promoter region.
Materials and methods
Patient 1 is a 43-year-old woman who was diagnosed with von Hippel–Lindau disease because of the presence of a retinal angioma and a cerebellar hemangioblastoma. We performed a complete molecular analysis of the VHL gene on genomic DNA of the proband and her asymptomatic 44-year-old brother and 31-year-old sister. The male individual tested negative while the sister was found to harbor the same alteration as in the proband. A magnetic resonance scan in this latter subject revealed the presence of a spinal hemangioblastoma.
Patient 2 is a 40-year-old male affected by cerebellar hemangioblastoma and renal oncocytoma. Patient 3 is a 12-year-old boy affected by isolated hemangioblastoma of the central nervous system. Patient 4 is a 16-year-old girl referred to our center for VHL testing due to an isolated retinal angioma. Family history in these latter three patients was unremarkable.
Signed informed consent prior to VHL molecular analysis was obtained from each tested individual.
Genomic DNA analysis
Genomic DNA was extracted from peripheral blood lymphocytes using standard protocols. VHL gene dosage was evaluated by quantitative Southern blot or real-time quantitative PCR analysis, as described (Stolle et al. 1998; Casarin et al. 2006). The coding region of the VHL gene was studied by direct sequencing as described (Murgia et al. 2000). PCR fragments obtained using primers 1AF (5′ GAAATACAGTAACGAGTTGGCCTAGC 3′) and 1AR (5′ ACCTCGGCCTCGTCCCAGT 3′) containing the identified −65_−55dup11 variant were separated on a 2.5% agarose gel. Individual bands were excised and DNA was recovered using a Qiaquick Gel extraction kit (Qiagen, Hilden Germany). The purified amplicon with the 11 bp insertion was cloned with a TOPO TA Cloning kit (Invitrogen, Carlsbad, CA) and sequenced using T7 primers according to the supplier’s protocol as previously described (Salviati et al. 2004).
mRNA analysis
Total mRNA was extracted from peripheral blood lymphocytes using RNA-Bee (TEL-TEST, Friendswood, TX) according to the manufacturer’s protocol. cDNA was synthesized using a Superscript II kit (Invitrogen) and random hexamers, according to the manufacturer’s protocol, and then amplified using primers 1CF (5′ GTGCTGCGCTCGGTGAACTC 3′) and 3R (5′ CAAGACTCATCAGTACCATCAAAAGCTG 3′). PCR conditions were as follows: 94°C 12 min; (94°C, 1 min; 63°C, 1 min; 72°C, 1 min) 40 cycles; 72°C, 10 min. PCR products were separated on a 10% acrylamide gel and visualized by silver staining. An aliquot of the PCR products was also separated in a 2% agarose gel. Bands were excised, purified as above, then sequenced using both amplification primers, and primer 2ex F (5′ AGGTCACCTTTGGCTCTTCAGA 3′) specific for the full-length VHL transcript.
PCR-RFLP analysis
The c.462 A > C variant was studied by means of a nested PCR protocol. An aliquot of the cDNA fragment, amplified with primers 1CF and 3R, and a genomic amplicon were re-amplified with primers P154Pmis F (5′ GACAGCCTATTTTTTGCCAATATCACACTGGC 3′) and either 3R (cDNA) or 2R (genomic DNA). A mismatched nucleotide in primer P154Pmis F creates a HaeIII site in the mutant allele. PCR conditions were: 94°C, 12 min; (94°C, 1 min; 63°C, 1 min; 72°C, 1 min) 15 cycles; 72°C, 7 min. PCR fragments digested by HaeIII (Roche, Basel, Switzerland), were separated on a 12% acrylamide gel, and visualized by silver staining. The c.1149 A/G polymorphism was assayed as described (Gläsker et al. 2001).
Results
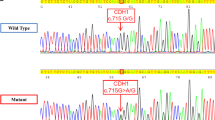
PCR–SSCP and direct sequencing of the VHL gene of patient 1 showed a heterozygous A>C nucleotide change at position c.462 of exon 2 (Fig. 1a). This substitution does not alter the sense of the corresponding codon (CCA>CCC, Pro>Pro). It was previously listed as a variant of unknown biological significance although predicted in silico to possibly disturb the splicing process of the gene (Olschwang et al. 1998). The mutation was present in the affected sister and absent in unaffected family members, as well as in 100 healthy controls. We detected no other nucleotide change in the affected individuals, and quantitative evaluation of the VHL gene dosage was also normal, excluding the possible presence of deletions undetected by conventional PCR techniques.

a Sequence of VHL exon 2 from genomic DNA of patient 1; the arrow indicates nt c.462. b VHL cDNA fragments amplified by RT-PCR. Lanes: M Molecular weight marker (pBR322/HaeIII), P proband, C normal control, B blank. c Sequence of VHL exon 2 from cDNA of patient 1; the arrow indicates nt c.462. d PCR–RFLP analysis (by Hae III digestion) of VHL exon 2 products amplified from genomic DNA and cDNA. Lanes: M Molecular weight marker (pBR322/HaeIII);1, 2 undigested and digested genomic DNA of patient 1; 3, 4 undigested and digested genomic DNA of negative control sample; 5, 6 undigested and digested cDNA of patient 1; 7, 8 undigested and digested cDNA of negative control sample
This substitution, however, affects the AG splicing consensus site at the 3′ end of exon 2. In order to demonstrate the presence of a splicing alteration, we amplified VHL cDNA from the patient’s peripheral blood lymphocytes. Two different transcripts of the VHL gene are normally present: a full-length transcript comprising exons 1, 2 and 3, and a shorter one, comprising only exons 1 and 3, the physiological significance of which is still unknown (Latif et al. 1993). The patient expressed both mRNAs and no abnormal transcripts were noted; however, the relative abundance of the full length form was apparently reduced in comparison to controls (Fig. 1b). Furthermore, sequencing of the PCR fragment demonstrated that the full-length transcript in the affected subject derived only from the wild-type allele (Fig. 1c). To confirm these data, and increase our sensitivity in detecting even small amounts of the mutant allele, we performed PCR–RFLP analysis of the mutation in both genomic and cDNA. The mutant allele was found in genomic DNA, while it was totally absent from the cDNA (Fig. 1d), indicating that the full length VHL transcript was expressed only from the wild type allele.
Molecular analysis of the VHL gene in patients 2, 3, and 4 did not detect pathogenic mutations in the coding region of the gene or in intron–exon boundaries, but revealed the presence of a duplication of 11 bp in the promoter region (c.−65_−55dup11) (Fig. 2a–c). Quantitative Southern blot/real-time quantitative PCR analyses were normal in these three patients, who had all inherited the duplication from a nonsymptomatic parent. Two siblings of patient 2, aged over 40 and also carriers of the same variant, underwent a thorough clinical evaluation, which was judged as negative. This variant was found in 1/100 control individuals.

a Sequence analysis of the VHL promoter region in patient 4. b Cloned wild-type allele. c Cloned duplicated allele; the 11 duplicated nucleotides are underlined. d PCR-RFLP analysis (by AccI digestion) of the VHL c.1149 A/G polymorphism on cDNA and genomic DNA. Lanes: M Molecular weight marker (pBR322/HaeIII), P patient 4, C control heterozygous individual, U uncut fragment
Nevertheless, the 11 bp duplication is located within the promoter region of the VHL gene, in the vicinity of the main transcription initiation site (Kuzmin et al. 1995), and within the putative binding site of several transcription factors (Zatyka et al. 2002). We therefore investigated the possibility that the variant could alter transcription of the gene from the affected allele.
Patient 4 was informative for a known polymorphic variant on the 3′UTR of the VHL gene (c.1149 A/G). As in the characterization of the mutation in patient 1, we took advantage of the presence of a sequence variant to discriminate expression of individual VHL alleles using a PCR–RFLP assay. Although not precisely quantitative, this approach showed that both alleles were expressed in apparently equimolar amounts (Fig. 2d) suggesting that the 11 bp duplication has no effect on expression of the affected allele.
Discussion
It is often difficult to establish pathogenicity of novel sequence variants discovered during mutational analysis in affected individuals. We have utilised a simple strategy to study the effect of two sequence variants on the pattern of transcription of theVHL gene.
Our data show that c.462 A > C is a pathogenic mutation affecting splicing of the VHL transcript. Several sequences within exons are recognized by the spliceosomal machinery, in particular, the last two nucleotides at the 3′ end of exons are essential in the splice donor consensus sequence (Cartegni et al. 2002). This mutation does not change the sense of the codon involved (both CCA and CCC encode proline), but it disrupts the splicing consensus at the 3′ end of exon 2. The affected mRNA cannot undergo correct maturation, and in fact, in our patient, full-length transcripts encoding active VHL protein derive only from the wild type allele. The consequences of this mutation on mRNA maturation are particularly severe; some splice-site mutations still allow the synthesis of small amounts of the normal transcript from the mutated allele (Moller et al. 2000), while in our case this seems to be completely abolished.
The second variant, in contrast, appears as a neutral polymorphism. Despite the fact that the duplication involves a critical region of the VHL promoter, it does not seem to have any appreciable impact on transcription of the affected allele. Although a subtle difference in the amount of the two transcripts cannot be excluded, our data indicate apparently equivalent levels of expression from both alleles. A strong argument in support of our hypothesis is the presence of the same variant in healthy relatives of our patients as well as in 1/100 control individuals.
Definition of the pathogenicity of a sequence variant is crucial in providing genetic counselling to probands and their families, as well as for their clinical management. We were in fact able to detect the presence of a spinal hemangioblastoma in the clinically asymptomatic sister of patient 1, who was also found to be carrier of the c.462 A>C mutation. On the other hand, we informed the relatives of individuals carrying the 11 bp duplication, that this is highly unlikely to be a pathogenic variant and is therefore not related to the tumors identified in the patients. It is interesting to note that the frequency of this variant has been found to be similar in the general population as in individuals referred to us for VHL-related tumors (1/100 vs 3/270).
We would like to draw attention to the importance of considering the potential pathogenic effects of sequence variants that may not belong to the obvious disease-causing mutation categories. Nucleotide variants within exonic sequences, even though not causing amino acid substitutions, may prove relevant for the proper function of splicing mechanisms; conversely, not all alterations in critical regulatory regions of a gene result in abnormal gene expression.
References
Cartegni L, Chew SL, Krainer AR (2002) Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet 3:285–298
Casarin A, Martella M, Polli R, Leonardi E, Anesi L, Murgia A (2006) Molecular characterization of large VHL deletions by quantitative real-time PCR: the hypothesis of an Alu-mediated mechanism underlying VHL gene rearrangements. Mol Diagn Ther 10:243–249
Gläsker S, Bender BU, Apel TW, van Velthoven V, Mulligan LM, Zentnerb J, Neumann HPH (2001) Reconsideration of biallelic inactivation of the VHL tumour suppressor gene in hemangioblastomas of the central nervous system. J Neurol Neurosurg Psychiatr 70:644–648
Kaelin WG, Maher ER (1998) The VHL tumour-suppressor gene paradigm. Trends Genet 14:423–426
Kuzmin I, Duh FM, Latif F, Geil L, Zbar B, Lerman MI (1995) Identification of the promoter of the human von Hippel–Lindau disease tumor suppressor gene. Oncogene 10:2185–2194
Latif F, Tory K, Gnarra J, Yao M, Duh F-M, Orcutt ML, Stackhouse T, Kuzmin I, Modi W, Geil L, Schmidt L, Zhou F, Li H, Wei MH, Chen F, Glenn G, Choyke P, Walther MM, Weng Y, Duan D-SR, Dean M, Glavac D, Richards FM, Crossey PA, Ferguson-Smith MA, Le Paslier D, Chumakov I, Cohen D, Chinault AC, Maher ER, Linehan WM, Zbar B, Lerman MI (1993) Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 260:1317–1320
Maher ER, Iselius L, Yates JRW, Littler M, Benjamin C, Harris R, Sampson J, Williams A, Ferguson-Smith MA, Morton N (1991) Von Hippel-Lindau disease: a genetic study. J Med Genet 28:443–447
Moller LB, Tumer Z, Lund C, Petersen C, Cole T, Hanusch R, Seidel J, Jensen LR, Horn N (2000) Similar splice-site mutations of the ATP7A gene lead to different phenotypes: classical Menkes disease or occipital horn syndrome. Am J Hum Genet 66:1211–1220
Murgia A, Martella M, Vinanzi C, Polli R, Perilongo G, Opocher G (2000) Somatic mosaicism in von Hippel–Lindau Disease. Hum Mutat 15:114
Olschwang S, Richard S, Boisson C, Giraud S, Laurent-Puig P, Resche F, Thomas G (1998) Germline mutation profile of the VHL gene in von Hippel–Lindau disease and in sporadic hemangioblastoma. Hum Mutat 12:424–430
Salviati L, Freehauf C, Sacconi S, DiMauro S, Thoma J, Tsai AC (2004) Novel SURF1 mutation in a child with subacute encephalopathy and without the radiological features of Leigh Syndrome. Am J Med Genet A 128:195–198
Stolle C, Glenn G, Zbar B, Humphrey JS, Choyke P, Walther M, Pack S, Hurley K, Andrey C, Klausner R, Linehan WM (1998) Improved detection of germline mutations in the von Hippel–Lindau disease tumor suppressor gene. Hum Mutat 12:417–423
Zatyka M, Morrissey C, Kuzmin I, Lerman MI, Latif F, Richards FM, Maher ER (2002) Genetic and functional analysis of the von Hippel–Lindau (VHL) tumour suppressor gene promoter. J Med Genet 39:463–472
Author information
Authors and Affiliations
Corresponding author
Additional information
Maddalena Martella and Leonardo Salviati contributed equally to this work.
Rights and permissions
About this article
Cite this article
Martella, M., Salviati, L., Casarin, A. et al. Molecular analysis of two uncharacterized sequence variants of the VHL gene. J Hum Genet 51, 964–968 (2006). https://doi.org/10.1007/s10038-006-0054-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-006-0054-9
Keywords
This article is cited by
-
The von Hippel–Lindau tumour suppressor gene: uncovering the expression of the pVHL172 isoform
British Journal of Cancer (2015)
-
Is CFTR 621+3 A>G a cystic fibrosis causing mutation?
Journal of Human Genetics (2010)
-
An 11-bp duplication in the promoter region of the VHL gene in a patient with cerebellar hemangioblastoma and renal oncocytoma
Journal of Human Genetics (2007)
