
Abstract
Spinocerebellar ataxia type 14 (SCA14) is an autosomal dominant neurodegenerative disorder, first described in a Japanese family, showing linkage to chromosome 19q13.4-qter. Recently, mutations have been identified in the PRKCG gene in families with SCA14. The PRKCG gene encodes the protein kinase Cγ (PKCγ), a member of a serine/threonine kinase family involved in signal transduction important for several cellular processes, including cell proliferation and synaptic transmission. To identify the disease-causing mutation in a large group of ataxia patients, we searched for mutations in the PRKCG gene. We ascertained 366 unrelated patients with spinocerebellar ataxia, either pure or with associated features such as epilepsy, mental retardation, seizures, paraplegia, and tremor. A C-to-G transversion in exon 4, resulting in a histidine-to-glutamine change at codon 101 of the PKCγ protein, was identified in patients from a family with slowly progressive pure cerebellar ataxia. Functional studies performed in HEK293 cells transfected with normal or mutant construct showed that this mutation affects PKCγ stability or solubility, verified by time-dependent decreased protein levels in cell culture. In conclusion, the H101Q mutation causes slowly progressive uncomplicated ataxia by interfering with PKCγ stability or solubility, which consequently may cause in either case a decrease in the overall PKCγ-dependent phosphorylation.
Similar content being viewed by others


Introduction
The spinocerebellar ataxias (SCAs) are a clinically and genetically highly heterogeneous group of inherited neurodegenerative disorders. They are characterized by progressive motor incoordination affecting the limbs and gait and are associated with degeneration of cerebellar Purkinje cells and brainstem neurons. Eleven SCA-associated genes have been identified to date, allowing the classification of these SCAs according to genotype and pathogenic mechanism (Schols et al. 2004). In SCA1, 2, Machado–Joseph disease (MJD/SCA3), 6, 7, 17, and dentatorubropallidoluysian atrophy (DRPLA), an expansion of a CAG repeat located within the coding region of the disease gene gives rise to an expanded polyglutamine tract in the disease protein. In SCA8, 10, and 12, the mutation consists in a tri- or pentanucleotide repeat expansion in a noncoding region. Alterations in amino acid composition, conducting to a deregulation in protein function, have been found to be responsible for cerebellar ataxia in SCA6, SCA14, and FGF14-related cerebellar ataxia (Schols et al. 2004).
Spinocerebellar ataxia type 14 (SCA14; MIM 605361) is an autosomal dominant disorder, first described in a three-generation Japanese family, showing linkage to chromosome 19q13.4-qter. The disease in this family was characterized by pure cerebellar ataxia or by intermittent axial myoclonus, followed by ataxia, in early-onset cases (Yamashita et al. 2000). Brkanac et al. (2002) described a SCA family of English and Dutch descent whose disease also showed linkage to the same region of chromosome 19. A missense mutation in the PRKCG gene was identified in patients from this family, all presenting pure cerebellar ataxia without myoclonus (Chen et al. 2003). A Q127R change in protein kinase Cγ (PKCγ), encoded by the PRKCG gene, has been found as the causative mutation in the Japanese family first showing linkage to this locus (Yabe et al. 2003). All of these mutations are in the regulatory domain of PKCγ (Fig. 1). The first mutation in the catalytic domain, F643L, was reported in a French family with cerebellar ataxia, myoclonus, and cognitive impairment (Stevanin et al. 2004). To date, a total of 11 mutations have been found in the PRKCG gene, seven of them in exon 4 (Chen et al. 2005).

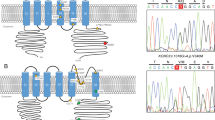
Primary structure of conventional PKC isozymes, showing the regulatory and catalytic domain. Conventional PKCs present a C1 domain that responds to diacylglycerol/phorbol esters, composed of two cystein-rich regions (Cys 1 and Cys 2) and a C2 domain that binds calcium ions. The catalytic domain contains two conserved phosphorylation sites: the turn motif (TM) and the hydrophobic motif (HM). The positions of the previously reported mutations, as well as the new mutation reported here (bold), are marked by arrows (Chen et al. 2003, 2005; van de Warrenburg et al. 2003; Yabe et al. 2003; Stevanin et al. 2004; Verbeek et al. 2005)
PKCγ is a member of a serine/threonine kinase family (Coussens et al. 1986; Knopf et al. 1986) involved in signal transduction relevant to several cellular processes, including cell proliferation and synaptic transmission (Tanaka and Nishizuka 1994; Zeidman et al. 1999). All PKCs have an amino-terminal regulatory domain as well as a carboxyl-terminal catalytic domain. They are grouped in three subclasses based on the composition of the regulatory domain (Mellor and Parker 1998). The two basic blocks of the regulatory domain, for conventional and novel PKCs, are C1 and C2, which are diacylglycerol and calcium sensors, respectively (Newton 2003), whereas the catalytic domain encloses kinase and substrate-recognition regions (Fig. 1).
Herein we report a Portuguese family with SCA14 presenting a mutation located in a highly conserved residue of the Cys 2 cystein-rich region of the amino-terminal regulatory domain, which causes downregulation of PKCγ.
Patients and methods
Subjects
We studied a total of 366 unrelated patients presenting SCA with or without associated symptoms such as epilepsy, mental retardation, seizures, paraplegia, and tremor. Among these patients, 103 represented families with apparently autosomal dominant inheritance, 41 were from families with patients in only one generation, and 222 were isolated cases; 325 unrelated affected subjects were of Portuguese origin, whereas 41 families were from Brazil. Repeat expansions responsible for SCA1, SCA2, MJD, SCA6, SCA7, SCA8, SCA10, SCA12, SCA17, and DRPLA were previously excluded in each of these 366 families (unpublished results).
Methods
Mutation analysis
Peripheral blood was collected from patients and their relatives after written informed consent was obtained. Genomic DNA was obtained from peripheral blood leukocytes by standard techniques (Sambrook et al. 1989).
Molecular analysis of exon 4 was performed by PCR amplification using the primers exon 4 forward (aaaagataaaagggcccctc) and reverse (aacccagaccgggcgctttg) (239 bp) and carried out with PCR conditions described elsewhere (Alonso et al. 2003). PCR products of exon 4 and intronic boundaries were screened for molecular variants by SSCP analysis (Orita et al. 1989). Electrophoresis was carried out in 15% polyacrylamide gels at 15°C. Conformational changes were confirmed by direct and reverse sequencing with BigDye Terminator Cycle Sequencing v1.1 Ready Reaction (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s instructions, and samples were analyzed on a 310 ABI PRISM genetic analyzer (Applied Biosystems).
cDNA constructs and site-directed mutagenesis
The human PRKCG full-length cDNA cloned in the pcDNA3/Amp vector was generously provided by Dr. S.M. Prescott (University of Utah). The C-to-G transversion found at the 303 position was introduced into the PRKCG cDNA using the QuickChange Site-Directed mutagenesis kit (Stratagene, La Jolla, CA, USA) according to the manufacturer’s instructions. The single base change was introduced using the following sense MDc303g-F (ccccggaacaaacagaagttccgcctgcatag) and antisense MDc303g-R (ctatgcaggcggaacttctgtttgttccgggg) oligonucleotides, which contained the mutant base. Successful mutagenesis was confirmed by sequencing normal and mutated constructs entirely (2,094 bp).
Cell culture and transfection
The HEK293 cells were propagated under standard conditions using Dulbecco’s modified Eagle’s medium with l-glutamine and 4,500 mg/l glucose and supplemented with 3.7 g/l sodium bicarbonate and 10% fetal bovine serum (Sigma, St. Louis, MO, USA). HEK293 cells were plated in 60-mm dishes. Cells were transfected with 5 μg of each expression vector encoding wild type (H101), mutant (Q101) PKCγ, or pcDNA3/Amp empty vector. All transfections were performed using the calcium phosphate precipitation method. Expression of the target protein was allowed for 36, 48, or 72 h.
RT-PCR
RNA extraction was performed with Trizol Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. RNA concentration was determined by spectrophotometry, and complementary DNA (cDNA) was generated from equal amounts of total RNA using ready-to-go RT-PCR beads (Amersham BioSciences, Piscataway, NJ, USA) according to the manufacturer’s instructions. Semiquantitative PCR was performed using, simultaneously, primers PKCγ-F (ccttctgcgaccactgt) and PKCγ-R (gctgcagttgtcagcat) (557 bp) to detect PKCγ, and ACTB–F (cgtcttcccctccatcgt) and ACTB–R (gcgcaagttaggttttgt) (1,096 bp) to detect β-actin, used as internal control. PCR was performed using the long template PCR system (Roche, Basel, Switzerland) under the manufacturer’s conditions: 0.15 μM of PKCγ-F and PKCγ-R primers, 1.5 μM of ACTB–F and ACTB–R primers, 350 μM dNTPs, expand long template system buffer 1 (17.5 mM MgCl2), and 4 U of expand long template enzyme mix, in a final volume of 50 μl; the PCR conditions consisted of an initial denaturing step at 95°C for 5 min, 30 cycles (95°C for 45 s, 55°C for 45 s, and 72°C for 1 min), and a final extension at 72°C for 5 min. Under these conditions, the exponential phase was determined for both genes, simultaneously, by PCR products accumulation at a constant rate. β-actin was also validated as an internal control, with a constant expression level across the different samples analyzed. Band quantifications were performed on the GS-800 calibrated imaging densitometer from Bio-Rad (Hercules, CA, USA).
Protein expression
Protein extraction was performed with RIPA buffer (150 mM NaCl, 50 mM Tris–HCl, 5 mM EGTA, 1% triton, 0.5% DOC, 0.1% SDS, pH 7.5) supplemented with phosphatase inhibitors (50 mM NaF, 1 mM Na3VO4 and 10 mM Na4PO7) in the presence of protease inhibitors (Roche).
Protein concentrations were determined using the Bio-Rad Dc (Hercules) assay according to the manufacturer’s protocol. Equal amounts (30 μg) of protein were loaded onto a 10% denaturing polyacrylamide gel and transferred to nitrocellulose membranes. Membranes were probed with anti-PKCγ (Transduction Laboratories, San Jose, CA, USA) and anti-β-actin, used as internal control (Sigma). The immunoreactive bands were visualized by the ECL Western blot detection kit (Amersham BioSciences). Band quantifications were performed on the GS-800 calibrated imaging densitometer from Bio-Rad (Hercules).
Statistical analysis
All quantitative data are expressed as mean ± SD of three independent experiments. Comparison of expression levels between wild type and mutant protein or mRNA was carried out using Student’s unpaired t-test.
Results
Mutation analysis
In one large Portuguese family, mutation detection revealed a mobility variant in exon 4, showing a three-band pattern (Fig. 2a). In the proband of this family, a C-to-G heterozygous transversion in nucleotide 303 of the ORF (GenBank accession number NM_002739) was detected by sequencing (Fig. 2b). This substitution produces a histidine-to-glutamine change at codon 101 of the PKCγ. Amino acid sequence alignment showed conservation of this His101 in the Cys 2 region among PKCγ isozymes from species such as M. musculus, R. norvegicus, O. cuniculus, B. taurus, D. melanogaster, and C. elegans, as well as in other human PKC isozymes (Fig. 3). Mutation analysis by SSCP did not detect this base change in 400 control chromosomes from the Portuguese population. All subjects from this family with DNA available were analyzed for the presence of this mutation (Fig. 2a). The mutation cosegregated with the disease in another affected individual (III10). Two asymptomatic at-risk subjects, aged 26 and 25, one with minor neurological signs, also inherited the mutation. Screening of exon 4, where the majority of SCA14 mutations are located, detected no mutations in the remaining 365 unrelated patients studied.

Pedigree of the family described, SSCP analysis, and sequencing of exon 4. a Black circles and squares indicate affected individuals with progressive cerebellar ataxia; a gray symbol indicates a subject referring no complaints but with abnormal signs on neurological exam; a dot in a symbol indicates an unaffected carrier. Patient IV9 refused to provide a sample for molecular analysis. Also shown are the disease ages at onset. PCR products were analyzed on 15% polyacrylamide gels by SSCP. A three-band pattern was detected in all patients with DNA available. b Sequencing of exon 4 in control and patient. A C-to-G substitution was detected at position 303, causing a histidine-to-glutamine change within the Cys 2 cystein-rich region of the PKCγ

Sequence alignments of PKCγ from different species and PKC isozymes. Residues from 90 to 136 of protein kinase Cγ correspond to the Cys 2 cystein-rich domain, showing the high degree of conservation of the mutated H101 position
Clinical findings
This family was characterized by pure cerebellar ataxia, with disease onset during or after the third decade and a benign course in the first two generations. In the younger generation the onset was earlier, in their 20s, and the clinical expression was also more severe. The proband of this family (III9), aged 65, had shown slowly progressive gait ataxia since the age of 30, followed by increasing difficulties in speaking and writing, and dysphagia by the age of 45. At examination, she presented moderate cerebellar dysarthria, dysphonia, bilateral horizontal nystagmus, limb dysmetria that was more severe in the lower limbs, and slight truncal and severe gait ataxia; no other neurological abnormalities were present. Her father (II3), who died at the age of 85 years, was said to have had gait imbalance since his fourth decade and to have been slightly dysarthric, as well having needed unilateral help walking in his later years. The only living affected brother of the proband (III10), aged 63, had gait ataxia with onset by the age of 30. His disease state had progressed slowly, with dysarthria by the age of 50 and dysphagia 10 years later. He was still able to walk unaided when he was last examined at age 63. Horizontal nystagmus and limb dysmetria were also observed at neurological examination. The proband’s niece (IV9), aged 35, first experienced unstable gait by the age of 26 years, followed by dysarthria 2 years later. She also had horizontal nystagmus and limb dysmetria. Although she was still able to walk without help for short distances, her neurological condition was clearly worse than her father’s. One of her siblings (IV10), still in his 20s, referred no complaints related to the disease, but he had horizontal nystagmus and slight heel-to-knee dysmetria and was unable to perform tandem walking.
Neuroimaging studies
Brain magnetic resonance imaging of the proband showed severe atrophy of the cerebellum, especially in the anterior part of the cerebellar hemispheres and in the superior vermis, as well as cerebral cortical atrophy (Fig. 4).

Brain magnetic resonance imaging of the proband (III9). a Saggital image showing severe atrophy of the cerebellum (arrow). b Coronal view showing superior vermis atrophy. c, d Axial image demonstrating atrophy in the anterior part of the cerebellar hemispheres
Functional studies
To investigate the influence of the H101Q mutation in PKCγ turnover, we compared PKCγ expression levels in HEK293 cells transfected with normal and mutant PKCγ. The levels of normal and mutant PRKCG mRNA did not significantly differ, showing that regulation of transcription is not altered and that transfection efficiencies were equal for normal and mutated constructs (Fig. 5a). A decrease in the expression level of the mutant protein was observed under the used conditions when compared with the normal. This effect increased with the time in culture after transfection, with a drop of 25%, 40%, and 60% at 36, 48, and 72 h of culture, respectively (Fig. 5b). These results were reproducible in three independent experiments.

PKCγ mRNA and protein expression in HEK293 cells. The cells were transfected with pcDNA3/Amp empty vector (Ø), pcDNA3/Amp with normal PKCγ construct (H101), or with pcDNA3/Amp with mutant PKCγ construct (Q101). a Left, normal levels of PKCγ mRNA detected in HEK293 cells by semiquantitative RT-PCR. Right, quantitative analysis of normal and mutant PKCγ mRNA levels; data are expressed as percentage of control. Means ± SD of three independent quantifications are shown. b Left, time-dependent reduction on mutant protein levels detected by Western blot analysis. Right, quantitative analysis of normal and mutant PKCγ protein levels; data are expressed as percentage of control. *P<0.05 from three independent experiments compared with normal protein levels. Means ± SD of the three experiments are shown
Discussion
In this study, we describe a new mutation in PKCγ, H101Q, causing SCA14 due to protein loss. By mutation analysis, we identified a new H101Q mutation in all available patients from a family with pure spinocerebellar ataxia. This mutation was not present in 400 control chromosomes. HEK293 cells expressing the mutant form of PKCγ (Q101) showed reduced PKCγ protein levels compared with normal PKCγ (H101). Although equal amounts of normal and mutant PRKCG mRNA were observed, reduction in mutant protein levels occurred with culture time, indicating that the mutant protein is putatively targeted for degradation at a higher rate. Alternatively, this result may be an indirect finding reflecting the protein insolubility or removal into cytoplasmic aggregates, as a previous report has shown that mutant PKCγ–GFP proteins readily aggregated in the cytoplasm, leading to cell death (Seki et al. 2005).
The PKCγ protein is composed of an amino-terminal regulatory domain and a carboxyl-terminal catalytic domain. The regulatory domain has two cystein-rich C1 domains (Cys 1 and Cys 2), each of which interacts with two zinc ions and makes available high-affinity binding sites for diacylglycerol and phorbol esters. The mutation H101Q, located in the Cys 2 cystein-rich region, replaces a residue critical for both zinc coordination and phorbol ester-binding (Fig. 1; Quest et al. 1994). Solution–structure modeling for a protein with a mutation that also replaces this residue, H101Y, showed that its conformation fluctuated widely, especially in the Zn1 and phorbol ester-binding sites (Chen et al. 2003). Our results show that a mutation at position H101 can interfere directly with protein function by affecting protein stability and probably by changing Zn2+ interaction and phorbol ester-binding, which may lead to loss of protein kinase activity. Two other mutations, G118D and C150F, also located on the regulatory domain of PKCγ, cause an increase in PKCγ kinase activity (Verbeek et al. 2005). Together with our results, this suggests that the mechanism underling SCA14 pathogenesis is triggered by changes in regulation of PKCγ. The altered phosphorylation of PKCγ substrates may be the first step in the disease mechanism, which is probably due to abnormalities in the innervation of Purkinje cells by climbing fibers, as observed in PKCγ knockout mice, which also suffer from abnormal gait and incoordination (Chen et al. 1995). It is thought that PKCγ is important in synaptic formation but not in early neuronal development; therefore, alterations in synaptic formation of Purkinje cells, caused by persistent multiple innervation of these cells by climbing fibers, may play an important role in disease associated with the H101Q mutation, as described here.
In this family, disease onset was during or after the third decade, with gait ataxia that progressed slowly and became associated with other cerebellar signs, such as dysphagia and dysarthria. The disease had a benign course in the first two generations but had an earlier onset in the youngest generation, with a more severe clinical expression. In the first family linked to the SCA14 locus (Yamashita et al. 2000), some patients presented cerebellar ataxia in the third decade of life, whereas those with an earlier onset began with myoclonus and only later developed cerebellar ataxia. The first families reported with PKCγ mutations in the regulatory domain had slowly progressive, uncomplicated cerebellar ataxia that began in the third or fourth decade of life, whereas patients with PKCγ mutations in the catalytic domain present a broader age at onset, ranging from childhood to the sixth decade of life (van de Warrenburg et al. 2003; Yabe et al. 2003; Stevanin et al. 2004; Chen et al. 2005).
The fact that SCA14 is an autosomal dominant disease suggests a pathogenetic mechanism based on gain of function of mutant proteins. Though immunohistochemical studies showed that Purkinje cells from a patient with H101Y stained very poorly for PKCγ (Chen et al. 2003), this together with the fact that aggregated PKCγ seems to be inactive (Seki et al. 2005) strongly supports a reduction of PKCγ in the etiology of SCA14. In this case, the mutant proteins could have a dominant negative effect by perhaps competing with the wild-type protein for substrate binding. The first disease-causing mutations identified in SCA genes were unstable tri- or pentanucleotide repeats, within coding or noncoding regions, that could lead to abnormalities in protein conformation or expression. The newly identified point mutations in PKCγ unveil a new perspective for understanding mutation effects on protein function and shedding light on the cellular pathways implicated in these diseases.
In conclusion, this new H101Q mutation, located on the Cys 2 cystein-rich region of protein kinase Cγ, causes a pure spinocerebellar ataxia, probably by interfering with protein stability or protein solubility, which in either case may lead to a decrease in the overall cell serine/threonine phosphorylation level.
References
Alonso I, Barros J, Tuna A, Coelho J, Sequeiros J, Silveira I, Coutinho P (2003) Phenotypes of spinocerebellar ataxia type 6 and familial hemiplegic migraine caused by a unique CACNA1A missense mutation in patients from a large family. Arch Neurol 60:610–614
Brkanac Z, Bylenok L, Fernandez M, Matsushita M, Lipe H, Wolff J, Nochlin D, Raskind WH, Bird TD (2002) A new dominant spinocerebellar ataxia linked to chromosome 19q13.4-qter. Arch Neurol 59:1291–1295
Chen C, Kano M, Abeliovich A, Chen L, Bao S, Kim JJ, Hashimoto K, Thompson RF, Tonegawa S (1995) Impaired motor coordination correlates with persistent multiple climbing fiber innervation in PKC gamma mutant mice. Cell 83:1233–1242
Chen DH, Brkanac Z, Verlinde CL, Tan XJ, Bylenok L, Nochlin D, Matsushita M, Lipe H, Wolff J, Fernandez M, Cimino PJ, Bird TD, Raskind WH (2003) Missense mutations in the regulatory domain of PKC gamma: a new mechanism for dominant nonepisodic cerebellar ataxia. Am J Hum Genet 72:839–849
Chen DH, Cimino PJ, Ranum LP, Zoghbi HY, Yabe I, Schut L, Margolis RL, Lipe HP, Feleke A, Matsushita M, Wolff J, Morgan C, Lau D, Fernandez M, Sasaki H, Raskind WH, Bird TD (2005) The clinical and genetic spectrum of spinocerebellar ataxia 14. Neurology 64:1258–1260
Coussens L, Parker PJ, Rhee L, Yang-Feng TL, Chen E, Waterfield MD, Francke U, Ullrich A (1986) Multiple, distinct forms of bovine and human protein kinase C suggest diversity in cellular signaling pathways. Science 233:859–866
Knopf JL, Lee MH, Sultzman LA, Kriz RW, Loomis CR, Hewick RM, Bell RM (1986) Cloning and expression of multiple protein kinase C cDNAs. Cell 46:491–502
Mellor H, Parker PJ (1998) The extended protein kinase C superfamily. Biochem J 332:281–292
Newton AC (2003) Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem J 370:361–371
Orita M, Suzuki Y, Sekiya T, Hayashi K (1989) Rapid and sensitive detection of point mutations and DNA polymorphisms using the polymerase chain reaction. Genomics 5:874–879
Quest AF, Bardes ES, Bell RM (1994) A phorbol ester binding domain of protein kinase C gamma. Deletion analysis of the Cys 2 domain defines a minimal 43-amino acid peptide. J Biol Chem 269:2961–2970
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
Schols L, Bauer P, Schmidt T, Schulte T, Riess O (2004) Autosomal dominant cerebellar ataxias: clinical features, genetics and pathogenesis. Lancet Neurol 3:291–304
Seki T, Adachi N, Ono Y, Mochizuki H, Hiramoto K, Amano T, Matsubayashi H, Matsumoto M, Kawakami H, Saito N, Sakai N (2005) Mutant protein kinase C gamma found in spinocerebellar ataxia type 14 is susceptible to aggregation and cause cell death. J Biol Chem 280(32):29096–29106
Stevanin G, Hahn V, Lohmann E, Bouslam N, Gouttard M, Soumphonphakdy C, Welter ML, Ollagnon-Roman E, Lemainque A, Ruberg M, Brice A, Durr A (2004) Mutation in the catalytic domain of protein kinase C gamma and extension of the phenotype associated with spinocerebellar ataxia type 14. Arch Neurol 61:1242–1248
Tanaka C, Nishizuka Y (1994) The protein kinase C family for neuronal signaling. Annu Rev Neurosci 17:551–567
van de Warrenburg BP, Verbeek DS, Piersma SJ, Hennekam FA, Pearson PL, Knoers NV, Kremer HP, Sinke RJ (2003) Identification of a novel SCA14 mutation in a Dutch autosomal dominant cerebellar ataxia family. Neurology 61:1760–1765
Verbeek DS, Knight MA, Harmison GG, Fischbeck KH, Howell BW (2005) Protein kinase C gamma mutations in spinocerebellar ataxia 14 increase kinase activity and alter membrane targeting. Brain 128:436–442
Yabe I, Sasaki H, Chen DH, Raskind WH, Bird TD, Yamashita I, Tsuji S, Kikuchi S, Tashiro K (2003) Spinocerebellar ataxia type 14 caused by a mutation in protein kinase C gamma. Arch Neurol 60:1749–1751
Yamashita I, Sasaki H, Yabe I, Fukazawa T, Nogoshi S, Komeichi K, Takada A, Shiraishi K, Takiyama Y, Nishizawa M, Kaneko J, Tanaka H, Tsuji S, Tashiro K (2000) A novel locus for dominant cerebellar ataxia (SCA14) maps to a 10.2-cM interval flanked by D19S206 and D19S605 on chromosome 19q13.4-qter. Ann Neurol 48:156–163
Zeidman R, Pettersson L, Sailaja PR, Truedsson E, Fagerstrom S, Pahlman S, Larsson C (1999) Novel and classical protein kinase C isoforms have different functions in proliferation, survival and differentiation of neuroblastoma cells. Int J Cancer 81:494–501
Acknowledgements
We thank all patients and their family members for cooperation, Prof. António Amorim for providing control DNA samples, and Nurse Helena Cardoso for collecting samples. We would also like to thank Dr. S.M. Prescott and Dr. M.K. Topham for kindly providing the PKCγ construct, Prof. Carlos Duarte for the suggestions and careful reading of this manuscript, and Victor Mendes for image technical assistance. This work was supported by research grants POCTI/MGI/34517/00, Financiamento Plurianual de Unidades de Investigação, all from FCT (Fundação para a Ciência e Tecnologia) and FEDER. I.A. and A.I.S. are recipients of scholarships from FCT, Portugal.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Alonso, I., Costa, C., Gomes, A. et al. A novel H101Q mutation causes PKCγ loss in spinocerebellar ataxia type 14. J Hum Genet 50, 523–529 (2005). https://doi.org/10.1007/s10038-005-0287-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-005-0287-z
Keywords
This article is cited by
-
Spinocerebellar ataxias (SCAs) caused by common mutations
neurogenetics (2021)
-
‘Medusa head ataxia’: the expanding spectrum of Purkinje cell antibodies in autoimmune cerebellar ataxia. Part 2: Anti-PKC-gamma, anti-GluR-delta2, anti-Ca/ARHGAP26 and anti-VGCC
Journal of Neuroinflammation (2015)
-
Spectrum and prevalence of autosomal dominant spinocerebellar ataxia in Hokkaido, the northern island of Japan: a study of 113 Japanese families
Journal of Human Genetics (2007)
-
Mutation of the highly conserved cysteine residue 131 of the SCA14 associated PRKCG gene in a family with slow progressive cerebellar ataxia
Journal of Neurology (2006)
