
Abstract
The 3’ rapid amplification of cDNA ends (3’ RACE) is widely used to isolate the cDNA of unknown 3’ flanking sequences. However, the conventional 3’ RACE often fails to amplify cDNA from a large transcript if there is a long distance between the 5’ gene-specific primer and poly(A) stretch, since the conventional 3’ RACE utilizes 3’ oligo-dT-containing primer complementary to the poly(A) tail of mRNA at the first strand cDNA synthesis. To overcome this problem, we have developed an improved 3’ RACE method suitable for the isolation of cDNA derived from very large transcripts. By using the oligonucleotide-containing random 9mer together with the GC-rich sequence for the suppression PCR technology at the first strand of cDNA synthesis, we have been able to amplify the cDNA from a very large transcript, such as the microtubule-actin crosslinking factor 1 (MACF1) gene, which codes a transcript of 20 kb in size. When there is no splicing variant, our highly specific amplification allows us to perform the direct sequencing of 3’ RACE products without requiring cloning in bacterial hosts. Thus, this stepwise 3’ RACE walking will help rapid characterization of the 3’ structure of a gene, even when it encodes a very large transcript.
Similar content being viewed by others
Introduction
Although the sequencing of the complete human genome revealed the presence of around 30,000–40,000 genes (Lander et al. 2001; Venter et al. 2001), it still remains a big challenge to determine the structure of every transcript due to the stage- and tissue-dependent nature of gene expression with complex splicing events associated with higher eukaryotes. Characterization of all splicing forms is essential to understand the function of each gene completely, since distinct splicing forms might have different functions (Rahman et al. 2002; Robertson and Jones 1999; Saitou et al. 2000). To determine the complete structure of each gene, isolation of cDNAs, including all splicing variants, is needed, either by screening the cDNA library or by the use of 5’ or 3’ rapid amplification of cDNA ends (5’ or 3’ RACE) (Frohman et al. 1988; Park et al. 2003). One of these, 3’ RACE, is widely used to isolate the cDNA of unknown 3’ flanking sequences. The conventional cDNAs are synthesized by oligo-dT priming, and 3’ RACE includes a simpler procedure that uses the naturally occurring poly(A) tail of mRNA as the common anchor site. However, 3’ RACE is not efficient when unknown coding regions are large, long 3’ untranslated regions are present, or when problematic template regions exist.
To overcome these problems, we have developed a method of high-performance, stepwise 3’ RACE walking for the structural analysis of large transcripts. In this method, cDNAs are synthesized using random 9mer flanked by a GC-rich sequence to allow suppression PCR technology (Chen et al. 1998; Park et al. 2003; Siebert et al. 1995). Because of the prominent specificity of amplification, the resultant PCR products can be sequenced directly without requiring cloning in bacterial hosts when there is no splicing variant.
To demonstrate applicability of our method, we have chosen the microtubule-actin crosslinking factor 1 (MACF1) gene (GenBank accession number AF317696) as a model system, since the size of its transcript is 20 kb (Byers et al. 1995; Gong et al. 2001) and it is impossible to perform 3’ RACE from the 5’ end of the transcript using the conventional method. Our method selectively amplified the region corresponding to the 5’ end of the MACF1 gene. Moreover, the PCR products can be used as a template for direct sequencing. Our method of stepwise 3’ RACE walking will help rapid characterization of the 3’ structure of a gene, even when it encodes a very large transcript.
Materials and methods
Oligonucleotide primer sequence
Oligonucleotide primer sequences are shown in Table 1.
RNA isolation and first-strand cDNA synthesis
Poly(A)+ RNA were isolated from Jurkat cell lines using MicroPoly(A)Pure (Ambion, Austin, TX, USA) and treated with DNA-free reagent (Ambion) to remove any residual contamination of genomic DNA by company-supplied protocol. The reverse transcription mixture contained 0.1 μg of heat denatured poly(A)+RNA, 2 or 10 pmol of random 9mer adaptor, 0.5 mM each of dNTP, 1.5 mM MgCl2, 37.5 mM KCl, 10 mM dithiothreitol, 15 U of RNase OUT (Invitrogen, Carlsbad, CA, USA), and 70 U of SuperScript III (Invitrogen) in 7.5 μl of 25 mM Tris-HCl pH 8.3. Incubation was done at 25°C for 10 min and then at 55°C for 1 h.
The 3’ RACE model of large transcripts
One twentieth of the cDNA was then used as a first PCR template with 3’ anchor primer 1 (AP1) and MACF1_0864U 5’ primer, followed by nested PCR using 3’ anchor primer 2 (AP2) and MACF1_0887U 5’ primer to enhance specificity. Amplification of each product was carried out using LA Taq (TaKaRa, Tokyo, Japan) according to company-supplied protocol: one cycle of denaturing at 95°C for 5 min, followed by 30–35 cycles of denaturing for 1 min at 95°C, annealing for 1 min at 55°C, and elongation for 3 min at 72°C. The PCR product was then analyzed using MACF1_0887U or MACF1_1138U 5’ primer by direct sequencing.
Results and discussion
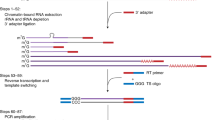
In conventional 3’ RACE, a cDNA synthesis primer containing oligo-dT and 3’ anchored PCR priming sites is used for the first-strand cDNA synthesis by the use of the naturally occurring poly(A) tail of mRNA as the common anchor site. Then first PCR and nested PCR are performed using two sets of 5’ gene-specific primers (GSPs) and 3’ APs to amplify the region between the gene-specific priming site and the poly(A) tail. However, this conventional 3’ RACE often fails when there is a large distance between GSP and AP due to the insufficient capability of reverse transcriptase and DNA polymerase. In contrast, as shown in Fig. 1A and B, our method utilizes random 9mer adaptor, oligonucleotide-containing random 9mer, and 3’ anchored PCR priming sites as a first-strand cDNA synthesis primer so that the cDNA can be synthesized from many different positions of mRNA, even close to the 5’ region of a very large transcript. Thus, the synthesized cDNAs are more likely to cover the entire transcript by overlapping each other. As a result, the distance between GSPs and APs is reduced enough to be amplified by ordinary PCR conditions. Moreover, by including suppression PCR technology (Chen et al. 1998; Park et al. 2003; Siebert et al. 1995), we were able to suppress amplification of nonspecific products; the templates possessing random 9mer adaptor on both ends formed an inverted repeat and built a form like a “panhandle” through the GC-rich region in the adaptor molecule (Fig. 1C). As the melting temperature of this structure was more than 85°C, it was more stable than the primer–template hybrid and therefore would suppress exponential amplification. When there was no splicing variation, we were able to analyze the structure of the PCR product by direct sequencing without cloning in bacterial hosts.

Schematic representation of the random 9mer adaptor 3’ rapid amplification of cDNA ends (RACE). A Synthesized cDNAs from different positions of mRNA. The synthesized cDNAs have more chance to cover the entire transcript by overlapping each other. B Model of stepwise random 9mer adaptor 3’ RACE. It identifies stepwise from 5’ to 3’ large transcript data. Dotted line indicates mRNA, black arrow indicates first strand cDNA produced in reverse transcription step, gray line indicates second strand cDNA products in first or nested PCR step, dotted box represents known sequence region, arrowheads indicate primers. GSP refers to 5’ gene-specific primer, AP refers to 3’ anchor primer. TTTT box indicates oligo-dT adaptor, (N) 9 box indicates random 9mer adaptor, Gppp indicates cap structure. C Suppression PCR technology. Remainder of random 9mer adaptor can anneal in second-strand synthesis during PCR. This synthesized strand readily forms a panhandle-like structure after the denaturing step because this structure is more stable than the primer-template hybrid and therefore suppresses exponential amplification and reduces nonspecific products. D PCR products from the 3’ RACE procedure. Size is shown in the right lane. The sample lanes depict products from synthesized cDNA templates of 10 pmol (lane 1) or 2 pmol (lane 2) random 9mer adaptor. There are correlations between random 9mer adaptor dose and length of PCR products. E Sequence analysis of each PCR product using direct sequencing. Chromatograms show dotted box position of each product. More sequence information of the 1.1 kb product is available than of the 0.2 kb and 0.7 kb products
As a model system, we performed 3’ RACE of the microtubule-actin crosslinking factor 1 (MACF1) gene from the region near the 5’ end of the transcript (Byers et al. 1995; Gong et al. 2001). Poly(A)+ RNA were isolated from Jurkat cell lines then treated with DNase to remove genomic contamination that would otherwise increase template complexity and the potential to yield an intronic rather than a exonic sequence. When we performed 3’ RACE with two sets of primers (AP1 and MACF1_0864U, and AP2 and MACF1_0887U for first PCR and nested PCR, respectively), we found the concentration of first-strand cDNA synthesis primer to be the most important factor in obtaining enough 3’ information of a given transcript. While using 10 pmol random 9mer adaptor in reverse transcription resulted in major products of 0.2 and 0.7 kb in size (Fig. 1D, lane 1), 2 pmol showed a major product of 1.1 kb (Fig. 1D, lane 2). Direct sequencing of these PCR products revealed that 0.2 and 0.7 kb bands were correctly derived from the sequence of the MACF1 gene at position 909–1559 base and also 1.1 kb band at position 909–1930 (Fig. 1E). We could then design the 5’ gene-specific primers for the next round of 3’ RACE walking from the obtained sequence. By repeating these steps four times, we were able to obtain a total of about 4 kb sequences from the 5’ region of the MACF1 transcript without cloning in a bacterial host (data not shown).
In reverse transcription, one molecule of mRNA annealed a few molecules of random 9mer adaptor in proportion to dose. Shown in Fig. 1D, lane 1 (10 pmol of random 9mer adaptor) products were smaller than those of lane 2 (2 pmol). Thus, it is assumed that annealing of many molecules of the random 9mer adaptor on one molecule of mRNA inhibits synthesis of long cDNAs. Since the length of synthesized cDNAs is dependent on the dose of random 9mer adaptor, concentration of random 9mer adaptor should be carefully adjusted to minimize the effort of 3’RACE walking.
CpG islands are believed to preferentially occur at the transcriptional start of genes, and it has been observed that most housekeeping genes have CpG islands at the 5’ end of the transcript (Gardiner-Garden and Frommer 1987; Lander et al. 2001; Larsen et al. 1992; Venter et al. 2001). Thus, our method can efficiently determine the structure of corresponding transcripts from only the 5’ end data around the CpG island. This method will be useful in the identification of genes or novel splicing variants.
References
Byers TJ, Beggs AH, McNally EM, Kunkel LM (1995) Novel actin crosslinker superfamily member identified by a two step degenerate PCR procedure. FEBS Lett 368:500–504
Chen SS, Chenchik A, Lukyanov KA, Siebert PD. (1998) Improved technique for walking in uncloned genomic DNA. In: Siebert PD, Larrick L (eds) Biotechniques Books. Natick, MA, pp 289–302
Frohman MA, Dush MK, Martin GR (1988) Rapid production of full-length cDNAs from rare transcripts: amplification using a single gene-specific oligonucleotide primer. Proc Natl Acad Sci 85:8998–9002
Gardiner-Garden M, Frommer M (1987) CpG islands in vertebrate genomes. J Mol Biol 196:261–282
Gong TW, Besirli CG, Lomax MI (2001) MACF1 gene structure: a hybrid of plectin and dystrophin. Mamm Genome 12:852–861
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al (2001) Initial sequencing and analysis of the human genome. Nature 409:860–921
Larsen F, Gundersen G, Lopez R, Prydz H (1992) CpG islands as gene markers in the human genome. Genomics 13:1095–1107
Park DJ, Pask AJ, Renfree MB, Graves JA (2003) 3’ RACE walking along a large cDNA employing tiered suppression PCR. Biotechniques 34:750–752, 754–756
Rahman L, Bliskovski V, Reinhold W, Zajac-Kaye M (2002) Alternative splicing of brain-specific PTB defines a tissue-specific isoform pattern that predicts distinct functional roles. Genomics 80:245–249
Robertson KD, Jones PA (1999) Tissue-specific alternative splicing in the human INK4a/ARF cell cycle regulatory locus. Oncogene 18:3810–3820
Saitou M, Sugimoto J, Hatakeyama T, Russo G, Isobe M (2000) Identification of the TCL6 genes within the breakpoint cluster region on chromosome 14q32 in T-cell leukemia. Oncogene 19:2796–2802
Siebert PD, Chenchik A, Kellogg DE, Lukyanov KA, Lukyanov SA (1995) An improved PCR method for walking in uncloned genomic DNA. Nucleic Acids Res 23:1087–1088
Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al (2001) The sequence of the human genome. Science 291:1304–1351
Acknowledgements
We thank Shawkat Haider for correction of English and critical reading of the manuscript. This research was partially supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports, and Culture of Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ozawa, T., Kondo, M. & Isobe, M. 3’ rapid amplification of cDNA ends (RACE) walking for rapid structural analysis of large transcripts. J Hum Genet 49, 102–105 (2004). https://doi.org/10.1007/s10038-003-0109-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-003-0109-0
