
Abstract
A mutation analysis of the BRCA1 gene in 140 French families with a history of breast cancer or breast-ovarian cancer revealed several deleterious germline mutations, as well as rare sequence variants. The 19 genetics variants were of 15 different types, two of which had not been reported in the Breast cancer Information Core (BIC) database. Five distinct truncating mutations, leading to putative nonfunctional proteins, were identified out of 140 index cases (3.5%). One novel nonsense mutation, C4491T, was reported, whereas the four other BRCA1 deleterious mutations identified consisted of frequent frameshifts in the nucleotide sequence. One splice variant (331+3A>G) and thirteen missense variations leading to amino acid substitutions of unknown structural and functional importance were identified. Among these, two BRCA1 missense mutations, A120G and T243C could be considered as suspected deleterious. The first missense mutation modified the initiation codon (M1V) and the second (C39R) may have consequences on the structure and functioning of the BRCA1 protein by modifying cysteine ligands from the RING finger domain. As expected BRCA1 gene alteration, including missense mutations of unknown biological significance, were more frequent in families with a history of breast-ovarian-cancer (32%) than in breast-cancer-only families (12%).
Similar content being viewed by others


Introduction
Breast cancer is a major cause of mortality by cancer in women (Parkin et al. 1999). Current estimations indicate that one in eight women will develop breast cancer during their lifetime. Approximately 5–10% of all breast cancers are thought to be due to an inherited predisposition (Claus et al. 1991). Some of these hereditary cases have been linked to mutations of the BRCA1 gene (Miki et al. 1994), a major breast cancer susceptibility gene. In fact, BRCA1 gene mutations are associated with 45% of breast cancers developed in these high-risk families (Easton et al. 1993). To date, more than 3,000 different genetic variants of BRCA1 have been described in the Breast cancer Information Core (BIC) database, which is accessible through the Web site hosted by the National Human Genome Research Institute at http://www.nhgri.nih.gov/Intramural_research/Lab_transfer/Bic. Novel genetic variants are constantly being identified, since, according to Szabo and co-workers, the number of mutation entries in the database increases at a steady rate of approximately 500 per year (Szabo et al. 2000).
In addition, it has been demonstrated that patients with hereditary breast cancer have a higher risk of developing contralateral breast cancer (Steinmann et al. 2001). Fortunately, different strategies for reducing this risk are available, including oophorectomy, mastectomy and chemoprevention (Narod et al. 2000). In this context, an alternative strategy would consist in identifying patients with mutations in the BRCA1 gene in order to prevent incidences of breast cancer more successfully.
In this study, we have analyzed the presence of mutations in the BRCA1 gene in 140 French families with a history of breast cancer or breast-ovarian cancer. For this purpose, we used a screening strategy based on denaturing gradient gel electrophoresis (DDGE) analyses, known to allow the identification of more than 80% of mutations (Serova et al. 1997). The majority of the families tested originate from the southeast of France. Nineteen (13.5%) of them were found to have variants in the BRCA1 gene sequence, including mutations of unknown structural and functional importance. Five of the mutations identified gave rise to a truncated BRCA1 protein. Furthermore, the mutation analysis of the BRCA1 gene also revealed three genetic variants that could be considered as deleterious.
Patients and methods
Patients
In 1992, a study was performed of hereditary breast cancer in southeastern France. From January 1992 to December 2000, 831 patients were seen in the Oncogenetic Department of the Antoine Lacassagne Anti-cancer Center. Among them, a total of 290 (35%) probands were selected, according to the following criteria: (1) at least one case of breast cancer diagnosed before the age of thirty; (2) two first-degree relatives, at least one of whom was affected before or at the age of forty; (3) three first-degree relatives with breast cancer, with one before or at the age of forty; (4) at least two breast cancers and one (or more) ovarian cancer; (5) at least two ovarian cancers (Easton et al. 1993). All information concerning the family history of cancer was obtained directly from the patients. The 140 families eligible for this study consisted of consecutive unselected families with histologically confirmed breast cancer, who were selected from the 290 families. Each selected patient was asked to read and sign an informed-consent form prior to donating blood samples. When a mutation was identified, the patient was contacted again and genetic counseling was provided prior to confirming the result on a new independent sample.
Out of the 140 patients, 23 (16.5%) had two first-degree relatives with breast cancer, at least one of whom was affected before or at the age of forty, 80 (57%) had three first-degree relatives with breast cancer, one before or at the age of forty, and 37 (26.5%) had at least two breast cancers and one (or more) ovarian cancer.
BRCA1 mutational analysis
From 20-ml blood samples, white blood cells were isolated and DNA extracted by the standard phenol-chloroform method.
All patients were analyzed for constitutional mutations throughout the BRCA1 gene by denaturing gradient gel electrophoresis (DGGE) (Fischer and Lerman 1983). All conditions, oligonucleotide sequences, PCR conditions electrophoretic conditions were obtained from Dr. Stoppa-Lyonnet of the Institut Curie, France (Stoppa-Lyonnet et al. 1997). All electrophoretic variants were verified by direct sequencing using a Perkin Elmer ABI 3700 automated sequencer according to the manufacturer's instructions.
All mutations and genetic variants were classified according to the BIC database website (http://www.nhgri.nih.gov/Intramural_research/Lab_transfer/Bic). The BIC database has adopted the nomenclature system recommendations of the HUGO Nomenclature Working Group (Antonarakis et al. 1998) for designation of mutations. The HUGO system recommends using the "A" of the ATG start codon as the number 1 position.
Mutations were interpreted as deleterious if they prematurely terminate the protein product and if some available evidences indicated a reasonable presumption of deleterious effect on protein function. Genetic variants of unknown significant biological effect includes mutations that occur in analyzed intronic regions whose clinical significance has not yet been determined. Whereas no deleterious mutations or polymorphism are defined as genetics variants for which published data demonstrate absence of substantial clinical significance or no significant effect on the amino acid sequence and stability.
Results
The mutation analysis of BRCA1 revealed deleterious mutations, as well as rare sequence variants, as described in Table 1. The 19 mutations identified are of 15 different types, including mutations of unknown structural and functional importance. Two of these genetic variants had not been reported in the BIC database at the time the manuscript of this paper was submitted. Seven common polymorphisms were also reported.
Five distinct truncating mutations, leading to putative nonfunctional proteins, were identified out of 140 index cases (3.5%), including one nonsense mutation, C4491A, reported for the first time. Four of the five deleterious BRCA1 mutations consisted of frameshifts of two, three or eleven nucleotide deletions and one nucleotide insertion. Among these frameshifts, the 5382insC at exon 20 and 185delAG at exon 2 were the most common mutations reported in the BIC. Two of the three founder mutations were found in Ashkenazi Jews. Another mutation, namely 3600del11, was reported to be frequent in the population native from northeastern France (Fricker et al. 2000).
Thirteen missense variations leading to amino acid substitutions of unknown structural and functional importance were identified. However, two BRCA1 missense mutations, A120G and T243C, may be considered as suspected deleterious mutations. Indeed, the first missense mutation, M1V, affected the initiation codon of the BRCA1 protein and the second rare missense, C39R, may have a major effect on the structure and functioning of the BRCA1 protein by modifying cystein ligands from the RING finger domain.
One splice variant, 331+3A>G, of uncertain biological significance was reported. It could be considered as a suspected relevant mutation since it occurred at a splice donor site (IVS5+3A>G). Ten genetic variants were reported in the large exon 11; however, five of them represented one of the three types of neutral polymorphism (A1186G, G1184A, C2731T).
Among the 81 patients from families with a history of breast cancer only, ten genetic variants were found (12%), including mutations of unknown biological significance. In this group, there appears to be a correlation between the number of breast cancer cases and the frequency of detected BRCA1 mutations. More surprisingly, the bilateral breast cancer sub-group (n=10) showed a lower frequency of detected BRCA1 genetic variants (10%).
In families with a history of breast-ovarian cancer, the incidence of genetic variants was 32% (9/28), whereas no mutations were observed in populations with one or more instances of bilateral breast cancer and ovarian cancer.
Discussion
In this study, we reported the screening of 140 families with a history of breast and/or ovarian cancer from southeastern France for the presence of gene mutations in BRCA1.
Nineteen germline mutations were found, among which five resulted in the truncation of the BRCA1 protein and fourteen missense mutations of unknown structural and functional importance. Seven neutral polymorphisms were also reported. The relatively low number of deleterious mutations (3.5%) may be explained by the testing of sporadic cases in predisposed families resulting in false-negative tests, or by technical insufficiencies. Indeed, since the sensitivity of the mutation-detection methods was not perfect, some mutations may have remained undetected. To verify this hypothesis, the 140 index cases are re-analyzing by denaturing high performance liquid chromatography (DHLPC) system. For example, a deleterious mutation, 2080delA, was recently determined in the index case of C40 family (data not showed). This intermediate result confirms that DHPLC system is a more sensitive method than DGGE (Wagner et al. 1999) and that all index cases should be re-analyzed. Moreover, it has been demonstrated that large BRCA1 rearrangements, which may account for more than 9% of cases, escaped detection by the DGGE method (Gad et al. 2002). These observations may explain why germline mutations found in this study are less frequent than expected.
It is also probable that the incidence of BRCA1 gene mutation was initially overestimated. Indeed, some reports suggested that in the general population about one woman in 500 might carry a BRCA1 mutation (Easton et al. 1993), while more recent articles reveal that the incidence of BRCA1 gene mutation may vary between 1/1,000 and 1/4,000 women (Antoniou et al. 2001).
Finally, a proportion of non-BRCA1 familial breast cancer cases found in this study certainly included BRCA2 mutation carriers. Indeed, the BRCA2 gene is thought to be responsible for an estimated one-third of familial female breast cancer and the majority of hereditary male breast cancer (Ford and Easton 1995). BRCA2 gene mutations of non-BRCA1 familial breast cancer cases determined in this study are under investigation.
However, the BRCA1 and BRCA2 genes do not explain all familial breast cancers. Thus, the large group of breast and ovarian cancer families, in which no BRCA1 and BRCA2 mutations were found, may represent an interesting group to investigate the involvement of a third gene in the development of breast and/or ovarian cancer in these families (Thompson et al. 2002).
In the BRCA1 mutation carriers, a higher incidence of genetic variants, including splice, missense and truncating mutations, is observed in families with a history of breast-ovarian cancer (32%) compared with families with a history of breast cancer only (12%). More surprisingly, the bilateral breast cancer sub-group showed a lower frequency of detected BRCA1 mutations (10%). It has been demonstrated that patients with hereditary breast cancer have a higher risk of contralateral breast cancer (Steinmann et al. 2001). However, the number of bilateral cases (n=10) screened in our work was probably insufficient compared with those observed in other studies.
The role in breast cancer susceptibility of the five missense mutations reported in the large exon 11 has been difficult to establish. However, a recent report demonstrated by a phylogenetic approach that some conserved regions of BRCA1 located at the 5′ and 3′ end of exon 11 are known to be involved in DNA double-stranded break repair (Fleming et al. 2003). Its conservation in species suggests that it is important for BRCA1 function. Therefore, one (S1040N) of the missense mutations reported in exon 11 that have been located in the RAD51-interacting domain (codons 758–1064) may influence BRCA1 function. However further studies should demonstrate by linkage analysis of high-risk families, functional assays or by demonstration of abnormal mRNA transcript processing that this genetic variant may be considered as deleterious. Indeed, the RAD51-interacting domain is large and probably a small number of amino acids may be crucial for BRCA1 function.
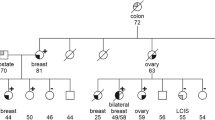
In this study, one splice variant, 331+3A>G, and two missense mutations of uncertain biological significance were reported as suspected to be deleterious. Indeed, the splice variant (IVS5+3A>G) occurred at a splice donor site, whereas the missense mutation A120G (M1V) modified the initiation codon of the BRCA1 protein. This evidence indicated reasonable presumptions that these two genetic variants could be considered as relevant mutations. The second BRCA1 missense mutation, suspected as deleterious, was a rare genetic variant, T243C (C39R), identified in a 53-year-old woman. Her family history was significant for its multiple cases of breast and ovarian cancers (Fig. 1). The case report has developed a breast cancer and a bilateral ovarian cancer. She was originally diagnosed with grade III epithelial carcinoma, a negative hormonal receptor status and no axillary nodal status. Two members of her family (two sisters of the mother's patient) developed breast cancer and died. The maternal grandmother died of breast-cancer-related causes. The identified missense mutation is located in one region of the BRCA1 sequence that encodes a 56-amino-acid sequence at the amino terminus, named the RING finger domain (Miki et al. 1994). Several pieces of data indicate that this zinc-binding motif, found in a variety of proteins, is important for the functioning of BRCA1 (Roehm et al. 1997). Therefore, a missense mutation that would modify cystein ligands from the RING finger domain may have an important action on the structure and functioning of the BRCA1 protein. This missense mutation, T243C, could be considered to severely disrupt the normal functioning of the BRCA1 protein and used as an indicator of the presence of an inherited mutation in her family.

Pedigree of a French family with the BRCA1 missense mutation T243C
Identifying BRCA1 mutations is important for counseling patients and deciding which treatment to use. Indeed, it has been demonstrated that prophylactic mastectomy reduces breast cancer incidence and mortality among groups with a high-risk family history, and women carrying BRCA1 mutations (Evans et al. 2001). Even if a recent report has shown that patients favored treatment and prevention options that involved minimal physical invasiveness (Cappelli et al. 2001), prophylactic oophorectomy should be offered to all women with BRCA1-BRCA2 mutations, especially those over childbearing age (Memarzadeh and Berek 2001). In addition, it has been demonstrated that tamoxifen, a selective estrogen receptor modulator, reduced the risk of contralateral breast cancer in women with pathogenic mutations in the BRCA1 or BRCA2 gene (Narod et al. 2000). The authors demonstrated that the protective effect of tamoxifen seems independent of that of oophorectomy.
It is therefore important to characterize breast cancer risk factors precisely in order to detect those women at most risk. In this way the prognosis of breast-conserving therapies, such as chemoprophylaxis or other actions, can be improved and the number of second cancers reduced.
References
Antonarakis SE (1998) Recommendations for a nomenclature system for human gene mutations. Nomenclature Working Group. Human Mutation 11:1–3
Antoniou AC, Pharoah PD, McMullan G, Day NE, Ponder BA, Easton D (2001) Evidence for further breast cancer susceptibility genes in addition to BRCA1 and BRCA2 in a population-based study. Genet Epidemiol 21:1–18
Cappelli M, Surh L, Humphreys L, Verma S, Logan D, Hunter A, Allanson J (2001) Measuring women's preferences for breast cancer treatments and BRCA1/BRCA2 testing. Qual Life Res 10:595–607
Claus EB, Risch N, Thompson WD (1991) Genetic analysis of breast cancer in the cancer and steroid hormone study. Am J Hum Genet 48:232–242
Easton DF, Bishop DT, Ford D, Crockford GP (1993) Genetic linkage analysis in familial breast and ovarian cancer: results from 214 families. The Breast Cancer Linkage Consortium. Am J Hum Genet 52:678–701
Evans D, Lalloo F, Shenton A, Boggis C, Howell A (2001) Uptake of screening and prevention in women at very high risk of breast cancer. Lancet 358:889–890
Fischer SG, Lerman LS (1983) DNA fragments differing by single base-pair substitutions are separated in denaturing gradient gels: correspondence with melting theory. Proc Natl Acad Sci USA 80:1579–1583
Fleming MA, Potter JD, Ramirez CJ, Ostrander GK, Ostrander EA (2003) Understanding missense mutations in the BRCA1 gene: an evolutionary approach. Proc Natl Acad Sci USA 100:1151–1156
Ford D, Easton DF (1995) The genetics of breast and ovarian cancer. Br J Cancer 72:805–812
Fricker JP, Muller D, Cutuli B, Rodier JF, Janser JC, Jung GM, Mors R, Petit T, Haegele P, Abecassis J (2000) Germ-line mutations of the BRCA1 gene in northeastern France. Bull Cancer 10:739–744
Gad S, Caux-Moncoutier V, Pages-Berhouet S, Gauthier-Villars M, Coupier I, Pujol P, Frenay M, Gilbert B, Maugard C, Bignon YJ, Chevrier A, Rossi A, Fricker JP, Nguyen TD, Demange L, Aurias A, Bensimon A, Stoppa-Lyonnet D (2002) Significant contribution of large BRCA1 gene rearrangements in 120 French breast and ovarian cancer families. Oncogene 21:6841–6847
Memarzadeh S, Berek JS (2001) Advances in the management of epithelial ovarian cancer. J Reprod Med 46:621–630
Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W (1994) A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 266:66–71
Narod SA (2001) Hormonal prevention of hereditary breast cancer. Ann N Y Acad Sci 952:36–43
Narod SA, Brunet JS, Ghadirian P, Robson M, Heimdal K, Neuhausen SL, Stoppa-Lyonnet D, Lerman C, Pasini B, de los Rios P, Weber B, Lynch H (2000) Tamoxifen and risk of contralateral breast cancer in BRCA1 and BRCA2 mutation carriers: a case-control study. Hereditary Breast Cancer Clinical Study Group. Lancet 356:1876–1881
Parkin DM, Pisani P, Ferlay J (1999) Global cancer statistics. CA Cancer J Clin 49:33–64
Roehm PC, Berg JM (1997) Sequential metal binding by the RING finger domain of BRCA1. Biochemistry 36:10240–10245
Serova OM, Mazoyer S, Puget N, Dubois V, Tonin P, Shugart YY, Goldgar D, Narod SA, Lynch HT, Lenoir GM (1997) Mutations in BRCA1 and BRCA2 in breast cancer families: are there more breast cancer-susceptibility genes? Am J Hum Genet 60:486–495
Steinmann D, Bremer M, Rades D, Skawran B, Siebrands C, Karstens JH, Dork T (2001) Mutations of the BRCA1 and BRCA2 genes in patients with bilateral breast cancer. Br J Cancer 85:850–858
Stoppa-Lyonnet D, Laurent-Puig P, Essioux L, Pages S, Ithier G, Ligot L, Fourquet A, Salmon RJ, Clough KB, Pouillart P, Bonaiti-Pellie C, Thomas G (1997) BRCA1 sequence variations in 160 individuals referred to a breast/ovarian family cancer clinic. Institut Curie Breast Cancer Group. Am J Hum Genet 60:1021–1030
Szabo C, Masiello A, Ryan JF, Brody LC (2000) The breast cancer information core: database design, structure, and scope. Hum Mutat 16:123–131
Thompson D, Szabo CI, Mangion J, Oldenburg RA, Odefrey F, Seal S, Barfoot R, Kroeze-Jansema K, Teare D, Rahman N, Renard H, Mann G, Hopper JL, Buys SS, Andrulis IL, Senie R, Daly MB, West D, Ostrander EA, Offit K, Peretz T, Osorio A, Benitez J, Nathanson KL, Sinilnikova OM, Olah E, Bignon YJ, Ruiz P, Badzioch MD, Vasen HF, Futreal AP, Phelan CM, Narod SA, Lynch HT, Ponder BA, Eeles RA, Meijers-Heijboer H, Stoppa-Lyonnet D, Couch FJ, Eccles DM, Evans DG, Chang-Claude J, Lenoir G, Weber BL, Devilee P, Easton DF, Goldgar DE, Stratton MR (2002) Evaluation of linkage of breast cancer to the putative BRCA3 locus on chromosome 13q21 in 128 multiple case families from the Breast Cancer Linkage Consortium. Proc Natl Acad Sci USA 99:827–831
Wagner T, Stoppa-Lyonnet D, Fleischmann E, Muhr D, Pages S, Sandberg T, Caux V, Moeslinger R, Langbauer G, Borg A, Oefner P (1999) Denaturing high-performance liquid chromatography detects reliably BRCA1 and BRCA2 mutations. Genomics 15:369–376
Acknowledgements
This work was supported by the Ligue Nationale Française Contre le Cancer, Comité Départemental des Alpes-Maritimes, and the Association pour la Recherche contre le Cancer. The authors wish to thank Dr. Dominique Stoppa-Lyonnet of the Institut Curie-France who has contributed to this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rostagno, P., Gioanni, J., Garino, E. et al. A mutation analysis of the BRCA1 gene in 140 families from southeast France with a history of breast and/or ovarian cancer. J Hum Genet 48, 362–366 (2003). https://doi.org/10.1007/s10038-003-0038-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-003-0038-y
