
Abstract
Tibial hemimelia is a rare congenital anomaly characterized by deficiency of the tibia with relatively intact fibula. Tibial hemimelia is identified as a solitary disorder, or a part of more complex malformation syndromes. Although the majority of cases with tibial hemimelia are sporadic, affected families with possible autosomal dominant or autosomal recessive inheritance have been reported. Here we report a pair of sibs, 6- and 2-year-old Japanese boys, with tibial hemimelia born to unrelated, phenotypically normal parents. The type of tibial hemimelia and associated malformations of hands and feet was quite different between the brothers. The elder brother was compatible with the Gollop–Wolfgang complex, and the younger brother with tibial agenesis–ectrodactyly syndrome. Screening of mutation by direct sequencing of candidate genes including Sonic hedgehog, HOXD-11, and HOXD-12 was unable to identify a disease-causing mutation.
Similar content being viewed by others


Introduction
Tibial hemimelia is a rare anomaly characterized by deficiency of the tibia with relatively intact fibula. It is classified into four types according to radiological criteria (Jones et al. 1978). The affected status of the tibia is variable, from complete to distal or proximal absence. Tibial hemimelia encompasses a heterogeneous group of disorders. It may present as an isolated anomaly, or may be associated with a variety of skeletal and extraskeletal malformations, such as polysyndactyly, clubhand, radioulnar synostosis, bifid femur, cleft lip/palate, and imperforate anus (Jones et al. 1978; Richieri-Costa et al. 1990). Tibial hemimelia may also constitute a part of more complicated malformation complex or syndrome, such as the Gollop–Wolfgang complex (Richieri-Costa et al. 1987a, 1987b; MIM: 228250), tibial agenesis–ectrodactyly syndrome (Majewski et al. 1985), and triphalangeal thumb–polysyndactyly syndrome (TPTPS: MIM 190605).
Tibial hemimelia is sporadic in the majority of cases; however, familial cases have been reported (Clark 1975; Majewski et al. 1985; Richieri-Costa et al. 1987a). In most reported families, the disorders follow an autosomal dominant pattern of inheritance, although they demonstrate great variability and reduced penetrance. In contrast, bilateral absence of tibia in three children from phenotypically normal parents has been described (Emami-Ahari and Mahloudji 1974). A child, born to healthy parents, who had tibial hemimelia and cleft lip/palate has also been reported (Richieri-Costa 1987). In both cases the parents are consanguineous, and thus an autosomal recessive inheritance is considered likely. Sib cases of tibial hemimelia to normal parents without consanguinity have also been reported (Wehbe et al. 1981; Mckay et al. 1984; Richieri-Costa et al. 1987a).
Identification of a responsible gene/genes would help to resolve these complex genetics of tibial hemimelia. Three related genes, Hoxd-11, HOXD-12, and Sonic hedgehog (SHH), can be a reasonable candidate. HOXD-12 affected pre- and post-axial chondrogenic branches in the limb and its misexpression in transgenic mice produced tibial hemimelia (Knezevic et al. 1997). HOXD-12 misexpression can induce ectopic Shh expression, resulting in mirror-image polydactyly (Knezevic et al. 1997). Shh is indispensable for limb skeleton formation as well as digit number and identity (Litingtung et al. 2002). Hoxd-11 misexpression in chick has produced a phenotype similar to transgenic Hoxd-12 (Morgan et al. 1992). Hoxd-11 up-regulates Shh expression in cultured limb cells, suggesting that it may participate in positive feedback regulation of Shh, also (Knezevic et al. 1997).
Here, we report a pair of sibs both having tibial hemimelia with a variety of associated malformations. They are products of phenotypically normal, unrelated parents. We performed mutation screening of SHH, HOXD-11, and HOXD-12 as candidate genes for the condition.
Patients and methods
Patients
The parents were healthy and not consanguineous, and had no other child. Their mother did not have diabetes mellitus nor had she taken any medicine during her pregnancy and had never experienced abortion. Detailed physical examination was performed on parents by expert orthopedists, but no malformation was found in hands, feet, or lower legs. There was no other person with malformation in their near relatives.
Patient 1
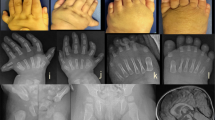
A 6-year-old elder brother was born at 38 weeks through a Cesarean section due to cephalo-pelvic disproportion. Birth weight was 3156 g. His intelligence was normal. His legs were both severely bowed with right tridactylous ectrodactyly and right femur process (Fig. 1A). His upper extremities were normal. He had no abdominal malformation. Roentgenogram demonstrated bilateral tibial hemimelia with intact fibula (Fig. 1B) and right bifid femur. According to Jones's classification (Jones et al. 1978), the right leg was classified to type Ia, and the left leg to type IV. Only the cuboid and three metatarsal bones and three phalanges were identified on the right foot. The left foot was normal. His right limb was amputated below the knee at the age of 1 year and 11 months, and his left limb was fixed between distal tibia and fibula with the ankle joint release at the age of 6 years.

Patient 1. A Photograph of the right leg at the age of 1 year and 11 months, showing tridactylous ectrodactyly and femoral process due to bifid femur. B Radiograph of lower extremities at 1 year and 9 months showing bilateral tibial hemimelia with intact fibula (arrows) and right bifid femur bone (arrow heads)
Patient 2
The younger brother, 2 years of age, was born by a prepared Cesarean section. Birth weight was 2800 g. A G-banding examination for amniotic fluid during pregnancy revealed no chromosome aberration. His intelligence was normal. He had no abdominal malformation. Both of his hands had four fingers. He did not have a triphalangeal thumb, however. His right leg was severely bowing with pre- and post-axial syndactyly of the toes; the left leg was normal (Fig. 2A). Roentgenogram demonstrated right tibial hemimelia with intact fibula corresponding to Jones's type II (Fig. 2B). The right hand had five metacarpals, but it had six proximal phalanxes, and second and third, fourth and fifth fingers were syndactylous, respectively; the left hand was ectrodactylous, lacking middle phalanges (Fig. 3). At the age of 1 year and 6 months, his right limb was fixed between remaining proximal tibia and fibula with ankle amputation and his right fingers were reconstructed.

Photograph (A) and radiograph (B) of patient 2. Lower extremities at 1 year and 5 months showing right six proximal phalanxes and syndactylous toes, and right tibial hemimelia (arrow) with intact fibula

Radiograph of patient 2. Hands at 1 year and 5 months showing polysyndactyly (right) and ectrodactyly (left)
Screening of mutation in candidate genes
Blood samples were obtained after written informed consent. Genomic DNA was extracted from peripheral blood leukocytes using standard procedures. Genomic sequences of SHH, HOXD-11, and HOXD-12 were obtained from GenBank (L38518, AF154915, AF154915). The entire coding regions the genes were amplified from genomic DNA using sets of primers (Table 1) that were designed using the Primer3 program (http://www.genome.wi.mit.edu/genome.software/other/pri-mer3.html).
PCRs were performed with the Takara exTaq and LA-Taq systems (Takara Shuzo, Otsu, Japan) according to the manufacturer's instruction using as templates 20 ng of genomic DNAs. PCR conditions were as follows: initial denaturation (94°C, 2 min) followed by 35–40 cycles of denaturation (94°C, 30 s), annealing (60°C according to Tm of the primers, 30 s), extension (72°C, 60 s), and final extension (72°C, 3 min). The PCR products were sequenced directly by means of an ABI Prism 3700 automated sequencer and the BigDye Terminator-Cycle Sequencing Kit (Applied Biosystems, Foster, CA).
Results and discussion
The phenotypes of the present sibs were different from each other in terms of tibial hemimelia and associated malformations (Table 2). The combination of bifurcation of the femur, tibial hemimelia, and ectrodactyly in the older brother was consistent with the Gollop–Wolfgang complex, whereas the phenotype in the younger brother was consistent with tibial agenesis–ectrodactyly syndrome. Affected families with phenotypic variations similar to those of the present sibs have previously been reported (Majewski et al., 1985; Richieri-Costa et al., 1987a, 1987b). Thus, the Gollop–Wolfgang complex and tibial agenesis–ectrodactyly syndrome can represent phenotypic variability of the same disorder.
The genetic basis of the present sibs remains elusive. The two sibs were derived from normal parents, which suggests autosomal recessive inheritance. An alternative explanation may be autosomal dominant inheritance with parental mosaicism (germinal and somatic) or reduced penetrance. Marked intra-familial phenotypic variation of tibial hemimelia has been reported (Yujnovsky et al. 1974; Majewsk et al. 1985; Canun 1984; Richieri-Costa et al. 1987a). More information is required to solve perplexing genetics of tibial hemimelia. Therefore, we screened mutation of candidate genes, SHH, HOXD-11, and HOXD-12. In the present sibs we did not find any mutations in the coding regions of these genes. However, these are still reasonable candidates for other patients with tibial hemimelia.
There are a few reports on the gene loci for tibial hemimelia. Kantaputra and Chalidapong (2000) reported a familial case of TPTPS associated with tibial hemimelia; the gene responsible for TPTPS was assigned to chromosome band 7q36 by linkage analysis (Tsukurov et al. 1994). A case of Langer–Giedion syndrome (LGS), which is assigned to 8q24, associated with tibial hemimelia has been reported (Stevens and Moore 1999). The authors proposed that the genes of the two conditions were contiguous. Turleau et al. (1982) also described a boy with LGS and bilateral tibial hemimelia. Future examination of these regions may lead to the identification of the disease gene for tibial hemimelia.
References
Canun S, Lomeli RM, Martinez R, Carnevale A (1984) Absent tibiae, triphalangeal thumbs and polydactyly: description of a family and prenatal diagnosis. Clin Genet 25(2):182–186
Clark MW (1975) Autosomal dominant inheritance of tibialmeromelia. J Bone Joint Surg Am 57(2):262–264
Emami-Ahari Z, Mahloudji M (1974) Bilateral absence of the tibias in three sibs. Birth Defects Orig Art Ser X(5):197–200
Jones D, Barnes J, Lloyd-Roberts GC (1978) Congenital aplasia and dysplasia of the tibia with intact fibula: classification and management. J Bone Joint Surg Br 60(1):31–39
Kantaputra PN, Chalidapong P (2000) Are triphalangeal thumb–polysyndactyly syndrome (TPTPS) and tibial hemimelia–polysyndactyly–triphalangeal thumb syndrome (THPTTS) identical? A father with TPTPS and his daughter with THPTTS in a Thai family. Am J Med Genet 93(2):126–131
Knezevic V, De Santo R, Schughart K, Huffstadt U, Chiang C, Mahon KA, Mackem S (1997) HOXD-12 differentially affects preaxial and postaxial chondrogenic branches in the limb and regulates Sonic hedgehog in a positive feedback loop. Development 124(22):4523–4536
Litingtung Y, Dahn RD, Li Y, Fallon JF, Chiang C (2002) Shh and Gli3 are dispensable for limb skeleton formation but regulate digit number and identity. Nature 418:979–983
Majewski F, Kuster W, ter Haar B, Goecke T (1985) Aplasia of tibia with split-hand/split-foot deformity: report of six families with 35 cases and considerations about variability and penetrance. Hum Genet 70(2):136–147
McKay M, Clarren SK, Zorn R (1984) Isolated tibial hemimelia in sibs: an autosomal-recessive disorder? Am J Med Genet 17(3):603–607
Morgan BA, Izpisua-Belmonte JC, Duboule D, Tabin CJ (1992) Targeted misexpression of Hox-4.6 in the avian limb bud causes apparent homeotic transformations. Nature 358:236–239
Richieri-Costa A (1987) Tibial hemimelia–cleft lip/palate in a Brazilian child born to consanguineous parents. Am J Med Genet 28:325–329
Richieri-Costa A, Ferrareto I, Masiero D, da Silva CRM (1987a) Tibial hemimelia: report on 37 new cases, clinical and genetic considerations. Am J Med Genet 27:867–884
Richieri-Costa A, Brunoni D, Laredo Filho J, Kasinski S (1987b) Tibial aplasia–ectrodactyly as variant expression of the Gollop–Wolfgang complex: report of a Brazilian family. Am J Med Genet 28:971–980
Richieri-Costa A, de Miranda E, Kamiya TY, Freire-Maia DV (1990) Autosomal dominant tibial hemimelia–polysyndactyly–triphalangeal thumbs syndrome: report of a Brazilian family. Am J Med Genet 36:1–6
Stevens CA, Moore CA (1999) Tibial hemimelia in Langer–Giedion syndrome: possible gene location for tibial hemimelia at 8q. Am J Med Genet 85(4):409–412
Tsukurov O, Boehmer A, Flynn J, Nicolai JP, Hamel BCJ, Traill S, Zaleske D, Mankin HJ, Yeon H, Ho C, Tabin C, Seidman C (1994) A complex bilateral polysyndactyly disease locus maps to chromosome 7q36. Nat Genet 6:282–286
Turleau C, Chavin-Colin F, de Grouchy J, Maroteaux P, Rivera H (1982) Langer–Giedion syndrome with and without del 8q: assignment of critical segment to 8q23. Hum Genet 62:183–187
Wehbe MA, Weinstein SL, Ponseti IV (1981) Tibial agenesis. J Pediatr Orthop 1(4):395–399
Yujnovsky O, Ayala D, Vincitorio A, Viale H, Sakati N, Nyhan WL (1974) A syndrome of polydactyly–syndactyly and triphalangeal thumbs in three generations. Clin Genet 6:51–59
Acknowledgements
We thank Dr Sen-ichi Tomizawa for help in performing the study. This work was supported by Grants-in Aids from the Ministry of Education, Culture, Sports and Science of Japan (contract grant number 14370476).
Author information
Authors and Affiliations
Corresponding author
Additional information
The first two authors contributed equally to this work.
Rights and permissions
About this article
Cite this article
Matsuyama, J., Mabuchi, A., Zhang, J. et al. A pair of sibs with tibial hemimelia born to phenotypically normal parents. J Hum Genet 48, 173–176 (2003). https://doi.org/10.1007/s10038-003-0003-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-003-0003-9
Keywords
This article is cited by
-
Fatty filum terminale and low-lying conus medullaris in Gollop-Wolfgang complex: a case report and review of literature
Child's Nervous System (2023)
-
Die kongenitale Tibiahemimelie
Der Orthopäde (2014)
-
Japanese founder duplications/triplications involving BHLHA9 are associated with split-hand/foot malformation with or without long bone deficiency and Gollop-Wolfgang complex
Orphanet Journal of Rare Diseases (2014)
-
Epidemiology, etiology, and genetic aspects of reduction deficiencies of the lower limb
Journal of Children's Orthopaedics (2008)
