
Abstract
Van der Knaap disease, or megalencephalic leukoencephalopathy with subcortical cysts (MLC), is an autosomal recessive disorder clinically characterized by macrocephaly, ataxia, spasticity, and mental decline. Magnetic resonance imaging (MRI) shows swollen brain with diffuse white-matter abnormalities and subcortical cysts, particularly in the anterior-temporal region. Recently, the MLC1 gene was identified as the gene responsible for this disorder, and mutations in this gene were described in several patients. We studied three Japanese patients with van der Knaap disease at the molecular genetic level. Two of them were homozygous for a previously-described mutation, S93L, and one was a compound heterozygote for S93L and a novel mutation, 452–468del+g, which leads to frameshift with a premature termination codon. Combining our data with previous reports allowed us to estimate the molecular genetic basis of this disorder in seven Japanese patients. In summary, S93L was observed in six of seven (85.7%) patients at least in one allele, and ten of 14 (71.4%) alleles had this mutation. Therefore, S93L appears to be fairly frequent in Japanese patients with van der Knaap disease, and analysis for this mutation in DNA isolated from leukocytes would provide for an easy and precise diagnosis of this disorder in Japanese patients.
Similar content being viewed by others

Introduction
Megalencephalic leukoencephalopathy with subcortical cysts (MLC; MIM604004), also called van der Knaap disease, is an autosomal recessive disorder characterized by macrocephaly, delayed onset of deterioration of motor functions with ataxia and spasticity, and mental decline (van der Knaap et al. 1995). Magnetic resonance imaging (MRI) shows swollen brain with diffuse white-matter abnormalities and subcortical cysts, particularly in the anterior-temporal region. A histopathological study revealed spongiform leukoencephalopathy without cortical involvement in this disorder (van der Knaap et al. 1996). A gene locus of this disorder was assigned on chromosome 22qtel (Topçu et al. 2000), the responsible gene, MLC1, was identified in the locus, and mutations in this gene were described in several patients (Leegwater et al. 2001). The function of the protein encoded by MLC1 is not yet known, but it is speculated to be a membrane protein with eight predicted transmembrane domains (Leegwater et al. 2001).
In this report, we have analyzed the MLC1 gene of three additional Japanese patients with van der Knaap disease and found a novel mutation and observed that S93L is quite common in Japanese patients.
Materials and methods
Patients and screening for S93L
All three patients studied are Japanese and apparently unrelated. Their clinical information is summarized in Table 1, and their MRIs are shown in Fig. 1. After informed consent was obtained from the parents of all three patients, peripheral leukocytes were collected. Initially, in order to detect a previously-described mutation, S93L (Leegwater et al. 2001), genomic DNA was isolated from peripheral leukocytes, and a genomic PCR fragment containing exon 4 was amplified by PCR with intronic primers 1 (5’-TTCAGATCCTTCTGGAAGCG-3’) and 2 (5’-ACACTGTCTGTCAGCCCCTC-3’). The PCR fragment was digested with the restriction endonuclease Sty I (New England Biolabs, Inc., Beverly, MA, USA), and electrophoresed through 3.0% NuSieve agarose (BMA, Rockland, ME, USA). The PCR product (210 bp) was cleaved into two segments (106 bp and 104 bp) in the presence of this nucleotide change, 393C>T (S93L), whereas the wild-type PCR product was not cleaved. The results were also confirmed by direct sequencing using the same primers.

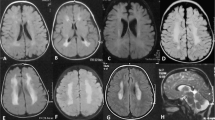
The brain MRI findings of the three patients. Patient 1 (a and b): a T2-weighted axial image shows diffuse hyperintensity in cerebral white matter (including subcortical U-fibers). b T1-weighted paramidsagittal image shows subcortical cysts in frontoparietal region. Patient 2 (c and d): c T2-weighted axial image demonstrates marked dilatation of lateral ventricles and diffuse hyperintensity in cerebral white matters. d FLAIR coronal image shows diffuse hyperintensity in cerebral white matter and focal hypointensity in the white matter of right inferior frontal gyrus, indicating a subcortical cyst. Patient 3 (e and f): e T2-weighted axial image shows macrocephaly and diffuse hyperintensity in cerebral white matters (including subcortical U-fibers). f FLAIR axial image shows diffuse hyperintensity of cerebral white matters and enlarged gyri accompanied with subcortical cyst in right temporal tip
Sequencing of the MLC1 cDNA
In the case of a patient who was not homozygous for S93L, then, total RNA was extracted from peripheral leukocytes using the Isogen kit (Nippon Gene, Tokyo, Japan). First-strand cDNA fragment synthesis was performed using oligo dT and the Superscript Preamplification System (Life Technologies, Rockville, MD, USA) following the manufacturer’s specifications. Three DNA fragments encompassing the entire coding region of the MLC1 cDNA were amplified by PCR using primers 3 (5’-ACACGTGGCTGTACATTCAG-3’) and 4 (5’-TGGGTTCAGGACTAGTTTGC-3’) for fragment 1, primers 5 (5’-ACGCCAATGTGATTCCCAAC-3’) and 6 (5’-AGACGTGAGGCTGCTTATGG-3’) for fragment 2, and primers 7 (5’-TGCCATTGCCAGTCATGTGG-3’) and 8 (5’-TTGTGCGTTTCCATGCTTGG-3’) for fragment 3. Our standard PCR conditions were: 30 cycles of denaturation at 94°C for 1 min; annealing at 65°C for 1 min; extension at 72°C for 2 min, and an additional extension at 72°C for 10 min. The PCR products were separated by electrophoresis through 1.5 % agarose gels, purified by GeneElute Agarose Spin Column (Sigma, St Louis, MO, USA), and directly sequenced using the BigDye Terminator Cycle Sequencing FS Ready Reaction kit using ABI Prism 310 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) according to manufacturer’s instructions.
Sequencing of the allele not having S93L
To determine the sequence of the other allele to that with S93L, primer 9 (5’-TCATTCCAGTGCATCCCTTC-3’) was designed, where underlined C is complementary only to the sequence of the wild-type (without S93L) allele and underlined T is mismatched exclusively to avoid misannealing to the allele. A genomic PCR fragment amplified using this primer was sequenced.
Results
Screening the three patients for S93L (393C>T) using PCR-RFLP revealed that two (patients 2 and 3) were homozygous and one (patient 1) was heterozygous for S93L (Fig. 2). To elucidate the other allele of patient 1, the MLC1 cDNA fragments were amplified by RT-PCR from leukocyte RNA of the patient and directly sequenced. The sequence was ambiguous and suggested the presence of a deletion or insertion in the other allele. Thus, we amplified only the unknown allele by genomic PCR using primer 9, which anneals to the allele not having S93L (see “Materials and methods”). As a result, we found a novel mutation, a deletion of 17-bp nucleotides, and a retention of one nucleotide g (452–468del+g) (Fig. 3a). No other nucleotide difference was found in the entire coding region of the cDNA between the patient and a control. Therefore, the patient was compound heterozygous for S93L and 452–468del+g. StyI digestion of a cDNA fragment amplified by RT-PCR using primers 3 and 4 showed both a cleaved fragment (with S93L) and an uncleaved fragment (with 452–468del+g), but the latter was much thinner than the former, implying that the amount of the mRNA containing 452–468del+g is reduced, probably because it is unstable (Fig. 3b, c).

Analysis for the S93L mutation. a Sequence of the MLC1 gene of patient 3 showing a C-to-T transition (S93L, arrow). b Representation of segments of the mutant PCR fragment and of the wild-type PCR fragment digested with Sty I. The numbers indicate segment lengths in base pairs. c Pedigree of patient 3 (Pt. 3) and electrophoresis on 3.0% NuSieve agarose of genomic PCR products of all patients (Pt. 1, patient 1; Pt. 2, patient 2) after digestion with Sty I. Fragment sizes (base pairs) are shown on the right

Analysis for the 452–468del+g mutation. a Sequence of a genomic PCR fragment specific for the allele without S93L of a control (upper) and of patient 1 (lower), showing 452–468del+g. b Representation of Sty I-digested segments of RT-PCR fragments (fragment 1) from the wild-type, the allele with 452–468del+g, and the allele with S93L. The numbers indicate segment lengths in base pairs. c Electrophoresis on 1.5% agarose of RT-PCR fragments of patient 1 and a control after digestion with Sty I. Note that the uncleaved band of patient 1 corresponds to the fragment from the allele with 452–468del+g. Fragment sizes (base pairs) are shown on both sides
Discussion
In this study, we analyzed the MLC1 gene in three newly identified Japanese patients. Two of them were homozygous for a previously-described mutation, S93L, and one was a compound heterozygote, with the S93L mutation on one allele and a novel mutation, 452–468del+g, which leads to a frameshift with a premature termination codon on the other allele. Together with results from a previous report (Leegwater et al. 2001) that described three Japanese patients who had three different mutations, S93L, S280L, and IVS11–2A>G, and with our previous report (Saijo et al. 2003) describing one Japanese patient who was homozygous for S93L, molecular genetic basis for this disorder has been established for a total of seven Japanese patients. In summary, S93L was observed six of seven (85.7%) patients at least in one allele, and ten of 14 (71.4%) alleles had this mutation. Therefore, S93L appears to be fairly frequent in Japanese patients with van der Knaap disease, and analysis for this mutation in DNA isolated from leukocytes would provide for an easy and precise diagnosis of this disorder in Japanese patients.
Although all the previous studies on the MLC1 gene used the genomic DNA from patients, we succeeded in effectively amplifying the MLC1 cDNA by RT-PCR from RNA extracted from leukocytes of patients in this study as well as a previous report (Saijo et al. 2003). While the protein encoded by the MLC1 gene may function mainly in the central nervous system, our results indicate that it is expressed in peripheral leukocytes at least sufficiently for RT-PCR. Since the MLC1 gene has 12 exons, our method using the cDNA simplifies the sequencing procedures as compared to the using genomic DNA.
While most patients who have been reported were below age 20 (van der Knaap et al. 1995; Topçu et al. 1998; Yalçinkaya et al. 2000; Ben-Zeev et al. 2001), the patients we describe are older: 23, 26, and 32 years for patients 1, 2, and 3 in this report, and 41 years for the patient we had previously reported (Saijo et al. 2003). This implies that, with appropriate care, patients with van der Knaap disease may have a good prognosis.
References
Ben-Zeev B, Gross V, Kushnir T, Shalev R, Hoffman C, Shinar Y, Pras E, Brand N (2001) Vacuolating megalencephalic leukoencephalopathy in 12 Israeli patients. J Child Neurol 16:93–99
Leegwater PAJ, Yuan BQ, van der Steen J, Mulders J, Könst AAM, Ilja Boor PK, Mejaski-Bosnjak V, van der Maarel SM, Frants RR, Oudejans CBM, Schutgens RBH, Pronk JC, van der Knaap MS (2001) Mutations of MLC1 (KIAA0027), encoding a putative membrane protein, cause megalencephalic leukoencephalopathy with subcortical cysts. Am J Hum Genet 68:831–838
Saijo H, Nakayama H, Ezoe T, Araki K, Sone S, Hamaguchi H, Suzuki H, Shiroma N, Kanazawa N, Tsujino S, Hirayama Y, Arima M (2003) A case of megalencephalic leukoencephalopathy with subcortical cysts (van der Knaap disease): molecular genetic study. Brain Dev 25:362–366
Topçu M, Saatci I, Topcuoglu MA, Kose G, Kunake B (1998) Megalencephaly and leukodystrophy with mild clinical course: a report on 12 new cases. Brain Dev 20:142–153
Topçu M, Gartioux C, Ribierre F, Yalçinkaya C, Tokus E, Oztekin N, Beckmann JS, Ozguc M, Seboun E (2000) Vacuoliting megalencephalic leukoencephalopathy with subcortical cysts, mapped to chromosome 22qtel. Am J Hum Genet 66:733–739
van der Knaap MS, Barth PG, Stroink H, van Nieuwenhuizen O, Arts WFM, Hoogenraad F, Valk J (1995) Leukoencephalopathy with swelling and a discrepantly mild clinical course in eight children. Ann Neurol 37:324–334
van der Knaap MS, Barth PG, Vrensen GFJM, Valk J (1996) Histopathology of an infantile-onset spongiform leukoencephalopathy with a discrepantly mild clinical course. Acta Neuropathol 92:206–212
Yalçinkaya C, Çomu S, Koçer N, Yuksel A, Gunduz E, Ocal A (2000) Siblings with cystic leukoencephalopathy and megalencephaly. J Child Neurol 15:690–693
Acknowledgements
We thank Dr. Fumihiko Sakai and Dr. Toyokazu Saito (Kitasato University) for their support, Dr. Naohide Shiroma (Ryukyu University) for useful discussion, Ms. Kazuko Endate for technical assistance, and Dr. Sara Shanske (Columbia University) for reviewing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tsujino, S., Kanazawa, N., Yoneyama, H. et al. A common mutation and a novel mutation in Japanese patients with van der Knaap disease. J Hum Genet 48, 605–608 (2003). https://doi.org/10.1007/s10038-003-0085-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-003-0085-4
Keywords
This article is cited by
-
Bipolar disorder in megalencephalic leukoencephalopathy with subcortical cysts: a case report
BMC Psychiatry (2020)
-
Megalencephalic leukoencephalopathy with cysts in twelve Egyptian patients: novel mutations in MLC1 and HEPACAM and a founder effect
Metabolic Brain Disease (2016)
-
MLC1 mutations in Japanese patients with megalencephalic leukoencephalopathy with subcortical cysts
Human Genome Variation (2014)
-
Identification of novel MLC1 mutations in Chinese patients with megalencephalic leukoencephalopathy with subcortical cysts (MLC)
Journal of Human Genetics (2011)
