
Abstract
Familial ependymal tumors are a very rare disease, the pathogenesis of which is unknown. Previous studies indicate an involvement of tumor suppressor genes localized within chromosomal region 22q, whereas details are still unclear. Here we report a non-neurofibromatosis type-2 (non-NF2) Japanese family in which two of the four members are affected with cervical spinal cord ependymoma, and one of the four is affected with schwannoma. Loss of heterozygosity (LOH) studies were carried out searching for common allelic loss at chromosomal region 22q11.2-qtel in two of the affected patients. Our findings support a prediction for existence of a tumor suppressor gene on chromosome 22 especially related to the tumorigenesis of familial ependymal tumors.
Similar content being viewed by others
Introduction
Ependymomas are rare glial tumors of the brain and spinal cord occurring both sporadically and in a familial syndrome. In the central nervous system they account for only 3–5% of all tumors, including those of childhood and, to a lesser extent, adult cases. In adults, over 60% of ependymomas arise in the spinal cord or filum terminale in the spine, and only 40% are intracranial (Read, 1884; Schiffer et al., 1991; West, 1985). Approximately 30% of ependymomas have monosomy 22. Also, cytogenetic studies have revealed a loss of chromosome 22 to be the most common genetic abnormality in ependymomas (Griffin et al., 1992; Ransom et al., 1992; Sainati et al., 1992), indicating an existence of important gene(s) within this region of the chromosome.
The neurofibromatosis type-2 gene (NF2) on chromosome 22q12 is responsible for type 2 neurofibromatosis syndrome, a hereditary neurocutaneous disorder that often develops glial tumors in the central nervous system, most often with ependymomas (Ruttledge et al., 1994). However, a few non-NF2 affected families with ependymomas have also been reported, although the number of cytogenetically investigated cases was not significantly large (Nijssen et al., 1994; Hulsebos et al., 1999). Contribution of a putative tumor suppressor gene(s) involved in the ependymoma tumorigenesis is still controversial.
We report here one Japanese family of non-NF2 familial spinal tumors, among whom two cases of spinal chord anaplastic ependymoma was detected with loss of heterozygosity at chromosome 22q, studied with the use of microsatellite markers.
Family and patients
Figure 1A shows the pedigree of the family. No signs of neurofibromatosis were present in this family.

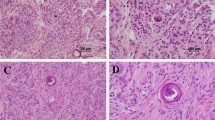
A A pedigree chart of the family. B magnetic response image of the neck of patient A. C microscopic appearance of an ependymoma illustrating the histopathology (hematoxylin and eosin-stained sections, ×40)
Patient A; born 1955 (33 years old disease onset). The patient visited an outpatient clinic of Tokyo Rousai Hospital complaining a sensory loss in both legs and the left hand, originally expanding from his left fingers, since 1988. Magnetic resonance imaging (MRI) on the cervical spine diagnosed an intramedullary cervical-cord tumor located at C1-C4 (Fig. 1B). Subtotal tumor resection was performed 1 month later, and a pathological diagnosis was made. The tumor was well vascularized and highly cellular with nuclear pleomorphism, several mitoses, and Rosette formation (Fig. 1C). Immunohistochemical staining revealed many GFAP-positive cells. In the electron microscopical photographs, cilia, microvilli, and desmosomes were identified on the tumor cells, indicating histological origin of these cells from ependymal cells. These findings led us to make a diagnosis of anaplastic ependymoma.
Patient B; born 1949 (44 years old at disease onset). The patient visited an outpatient clinic of Tokyo Rousai Hospital, complaining of muscle weakness on right-thumb motion since 1992. Within a year, progression of the weakness expanded into second to fourth finger flexion and extension. In the next year (1994), similar symptoms appeared on the left side of the hand. An MRI on the cervical and thoracic cord revealed an intramedullary thoracic-cord tumor located at C4-Th4. A subtotal tumor resection was performed. Pathohistological findings were similar to those of patient A, and thus the histological diagnosis of anaplastic ependymoma was made.
Patient C; born 1948 (45 years old at disease onset). The patient first visited the outpatient clinic of Tokyo Rousai Hospital on March 1994 complaining with neck dullness and uncomfortable feelings in his left arm that had been continuing for about 4 months prior to the visit. Although there were no apparent objective neurological findings, MRI revealed a cystic lesion in the paravertebral portion of lower cervical cord. Clinically, the tumor was diagnosed as the eighth cervical nerve root Shwannoma associated with cystic degeneration. Tumor resection was operated under the control of the department of orthopedics 6 months after the first visit. Pathological diagnosis was consistent with the clinical one.
Molecular genetics study
To make an LOH analysis of the family members using microsatellite markers, genomic DNA was extracted from blood leukocytes from two unaffected family members and from the formalin-fixed, paraffin-embedded tumor tissue from the affected members, as described previously (Kitamura et al., 2000). Microsatellite markers located at loci covering chromosome 22q were chosen, as listed in Table 1. Primer sequences and polymerase chain reaction (PCR) conditions were taken from the relevant databases by access though the Internet. The known cytogenetic localizations of markers and the order of markers on chromosome 22 used in this study were deduced from published maps (http://www.ncbi.nlm.nih.gov/genemap99; Dunham et al., 1999). Definition of LOH in the tumors was made by visual inspection of autoradiographs. However, because of heavily degraded DNA, the LOH status of 12 markers could hardly be successfully determined in the tumor of two patients (patient B and patient C).
Representative autoradiograms of two patients with deletions at chromosome 22q are shown in Fig. 2. Patient B and C showed retained alleles at D22S427 but showed LOH at D22S539, D22S1176, D22S1156, D22S1165, D22S922, and D22S1169 markers, which contain the NF2 gene in 22q11.2-qtel. However, allelic loss was not detected for the TP53 marker at the 17p13.1 or in the interval containing the DMBT1 locus between D10S214 and D10S1708 markers at 10q25-q26 (Table 1).

Autoradiographs showing the genotype analysis of family members and the loss of heterozygosity (LOH) analysis of the tumor of two patients. Locations and order of microsatellite markers (left) were derived from published linkage information; distances between loci are indicated in centimorgans. N and T indicate matched DNA samples isolated from normal and tumor tissues, respectively. ROH retention of heterozygosity, ND no detection
To search for germline mutation in the NF2 gene, we amplified all seventeen exons of the gene in genomic DNA from blood of patients B and C as well as cancer-free volunteers (Harada et al, 2001) using the primers described by Jacoby et al. (1994) (Table 2). Direct sequencing of the entire coding region was carried out as previously described (Iwasaki et al, 2001). No germline mutations of the NF2 gene were found in either patient (data not shown).
Discussion
The present study has shown a loss of chromosomal region in 22q in a non-NF2 family with ependymoma, supporting a recently proposed perspective for a pathogenesis of rare familial ependymomas (Bijlsma et al., 1995; Ransom et al., 1992; Weremowicz et al., 1992; Wernicke et al., 1995). Although we could not define the exact locus of the responsible gene in this study, our results suggest that 22q deletions could play an important role in the pathogenesis of ependymomas.
Familial ependymoma is a rare disease, most of which are associated with type-2 neurofibromatosis. Because type-2 neurofibromatosis is caused by a single NF2 gene mutation, and their associated ependymoma usually represent a loss of heterozygosity in the NF2 gene locus on chromosome 22, significant contribution of this gene for the pathogenesis of ependymoma has been proposed. Those studies have reported several cytogenetic alterations in ependymomas, among which deletions or translocations involving chromosome 22q have been the most frequent changes (Bijlsma et al., 1995; Ransom et al., 1992; Weremowicz et al., 1992; Wernicke et al., 1995). However, there is considerable variability in the frequency of allelic losses involving chromosome 22q in various studies. Therefore, contribution of genes located within this chromosomal region is not yet clear. In addition, several other regions, including 17p13 and 10q25–26, have been proposed in some relationships between the pathogenesis of ependymomas.
A chromosome of great interest in the study of adult human cancer has been chromosome 17, primarily because of the location of the important tumor suppressor gene TP53 on 17p. Genetic study detected 17p DNA loss in nine of 18 pediatric ependymomas (Von Haken et al, 1996). Most of these DNA losses were distal to the p53 locus. Other studies have also shown that p53 gene mutations are infrequent in adult ependymomas (Ohgaki et al., 1991;Von Haken et al., 1996). Monosomy 10 also has been reported in a few cases of ependymomas (Weremowicz et al., 1992). The deletion was mapped to 10q25.3–26.1, a region previously assigned to deletions and rearrangements in glioblastoma multiform and other malignant tumors, and the recently isolated candidate tumor suppressor gene DMBT1, spanning this deletion (Mollenhauer J et al., 1997). Therefore, those possibilities should have been examined in our cases.
In fact, our study delineated all regions on chromosome 22q showing LOH (Fig. 2) in addition to other candidate loci. As expected, allelic loss was detected in the wide range of the portion containing the NF2 locus at the 22q11.2-qtel region in both patients. On the contrary, allelic loss was not detected for the TP53 marker at the 17p13.1 or in the interval containing the DMBT1 locus between D10S214 and D10S1708 markers at the 10q25-q26 (Table 1). Therefore, at least in our cases, contribution of gene(s) located within chromosome 22q was indicated.
Of course, the NF2 gene is a most probable candidate susceptibility gene located in the 22q12, whereas we could not make certain by defining the common LOH region or by searching for neutral mutation in this gene locus. Nevertheless, we assume a significant contribution of this gene for tumorigenesis of ependymomas. Alternatively, the other possibility that another gene located within 22q actually affected the tumorigenesis of ependymoma can not be excluded. In fact, LOH studies for extensively collected ependymoma families indicate that ependymoma tumor suppressor gene may also be present in region 22pter-22q11.2 (Hulsebos et al., 1999). Thus, the existence of a tumor suppressor gene on chromosome 22 other than the NF2 was speculated. These possibilities should be tested in future investigations.
In summary, this present study defined a candidate region of the ependymoma susceptibility gene by means of loss of heterozygosity tests in two patients of familial ependymoma unrelated to neurofibromatosis (between D22S539 to D22S1169 markers at 22q11.2-qtel). Our results suggest that there is at least one tumor suppressor gene on chromosome 22q. The consistent finding of loss of chromosome 22 in our cases and the other previously reported a few cases suggests a causal relation in the pathogenesis of familial ependymoma tumors.
References
Bijlsma EK, Voesten AM, Bijleveld EH, Troost D, Westerveld A, Merel P, Thomas G, Hulsebos TJ (1995) Molecular analysis of genetic changes in ependymomas. Genes Chromosomes Cancer 13:272–277
Dunham N, Shimizu BA, Roe S, Chissoe et al. (1999) The DNA sequence of human chromosome 22. Nature 402:489–495
Griffin CL, Carson BS, Brem H (1992) Chromosomal abnormalities in low-grade central nervous system tumors. Cancer Genet Cytogenet 60:67–73
Harada H, Nagai H, Tsuneizumi M, Mikami I, Sugano S, Emi M (2001) Identification of DMC1, a novel gene in the TOC region on 17q25.1 that shows loss of expression in multiple human cancers. J Hum Genet 46:90–95
Hulsebos TJ, Oskam NT, Bijleveld EH, Westerveld A, Hermsen MA, van den Ouweland AMW, Hamel BC, Tijssen CC (1999) Evidence for an ependymoma tumour suppressor gene in chromosome region 22pter-22q11.2. Br J Cancer 81:1150–1154
Iwasaki H, Ota N, Nakajima T, Shinohara Y, Kodaira M, Kajita M, Emi M (2001) Five novel single-nucleotide polymorphisms of human interferon gamma identified by sequencing the entire gene. J Hum Genet 46:32–34
Jacoby LB, MacCollin M, Louis DN, Mohney T, Rubio M-P, Pulaski K, Trofatter JA, Kley N, Selzlnger B, Ramesh V, Gusela JF (1994) Exon scanning for mutation of the NF2 gene in schwannomas. Hum Mol Genet. 3:413–419
Kitamura Y, Shimizu K, Tanaka S, Ito K, Emi M (2000) Association of allelic loss on 1q, 4p, 7q, 9p, 9q, and 16q with postoperative death in papillary thyroid carcinoma. Clin. Cancer Res 6:1819–1825
Mollenhauer J, Wiemann S, Scheurlen W, Korn B, Hayashi Y, Wilgenbus KK, von Deimling A, Poustka A (1997) DMBT1, a new member of the SRCR supertamily, on chromosome 10q25.3–26.1 is deleted in malignant brain tumours. Nature Genet 17:32–39
Nijssen PC, Deprez RH, Tijssen CC, Hagemeijer A, Arnoldus EP, Teepen JL, Holl R, Niermeyer MF (1994) Familial anaplastic ependymoma: evidence of loss of chromosome 22 in tumour cells. J Neurol Neurosurg Psychiatry 57:1245–1248
Ohgaki H, Eibl RH, Wiestier OD, Yasargil MG, Newcomb EW, Keihues P (1991) p53 mutations in nonastrotic human brain tumors. Cancer Res 51:6202–6205
Ransom DR, Kimmel DW, Moertel CA, Dahi RJ, Scheithauer BW, Kelly PJ, Jenkins RB (1992) Cytogenetic and loss of heterozygoity studies in ependymomas, pilocytic astrocytomas and oligodendrogliomas. Genes Chromosom Cancer 5:348–356
Read G (1984) The treatment of ependymoma of the brain or spinal canal by radiotherapy. Clin Radiol 35:163–166
Ruttledge MH, Sarrazin J, Rangaratnam S, Phelan CM, Twist E, Merel P, Delattre O, Thomas G, Nordenskjold M, Collins VP, Dumanski JP, Rouleau GA (1994) Evidence for the complete inactivation of the NF2 gene in the majority of sporadic meningiomas. Nat Genet 6:180–184
Sainati LM, Putti MG, Giangaspero F, Rigobello M, Stella M, Zanesco L, Basso G (1992) Cytogenetic t(11;17)(q13;q21) in a pediatric ependymoma. Cancer Genet Cytogenet 59:213–216
Schiffer D, Chio A, Cravioto H, Giordana MT, Migheli A, Soffietti R, Vigliani MC (1991) Ependymoma internal correlations among pathological signs: the anaplastic variant. Neurosurgery 29:206–210
von Haken MS, White EC, Daneshvar-Shyesther L, Sih S, Choi E, Kalra R, Cogen PH (1996) Molecular genetic analysis of chromosome arm 17p and chromosome arm 22q DNA sequences in sporadic pediatric ependymomas. Genes Chromosomes Cancer 17:37–44
Weremowicz S, Kupsky WJ, Morton CC, Fletcher JA (1992) Cytogenetic evidence for a chromosome 22 tumor suppressor gene in ependymoma. Cancer Genet Cytogenet 61:193–196
Wernicke C, Thiel G, Lozanova T, Vogel S, Kintzel D, Janisch W, Lehmann K, Witkowski R (1995) Involvement of chromosome 22 in ependymomas. Cancer Genet Cytogenet 79:173–176
West C (1985) Ependymomas: factors in clinical and diagnostic staging. Cancer 56:1812–1816
Acknowledgment
Supported by special grants-in-aid for cancer research and “Genome Science” from the Ministry of Education, Science, and Culture of Japan; and by a research grant for cancer research from the Ministry of Health and Welfare of Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yokota, T., Tachizawa, T., Fukino, K. et al. A family with spinal anaplastic ependymoma: evidence of loss of chromosome 22q in tumor. J Hum Genet 48, 598–602 (2003). https://doi.org/10.1007/s10038-003-0078-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-003-0078-3
Keywords
This article is cited by
-
Spinal cord ependymomas and the appearance of other de novo tumors: a systematic review
Journal of Medical Case Reports (2014)
