
Abstract
Endophytes, important plant-associated mycobionts, have attracted a great deal of attention because of their bioactive secondary metabolites. Even though halophytes have been reported to overcome salt stress via associations with their endophytes, few studies have investigated the metabolites produced by the endophytes from halophytes. In this study, a dark septate endophytic fungal strain (JS0464), identified as Gaeumannomyces sp. by ITS sequencing, was isolated from the rhizome of a halophyte, Phragmites communis, in Suncheon bay, South Korea. This strain was cultured on a large scale and extracted with ethyl acetate. Chemical investigations of extracts of JS0464 led to the isolation of two glycosylated dialkylresorcinol derivatives (1–2), an anthraquinone derivative (3) and eight known compounds (4–11), which were identified by spectroscopic analyses incorporating one-dimensional/2D NMR and MS. Nine compounds showed significant nitric oxide reduction activity in lipopolysaccharide-stimulated microglia BV-2 cells, seven of which did not impair cell viability. The results suggest that endophytes from the halophytes could be potential resources for bioactive natural products.
Similar content being viewed by others

Introduction
Endophytes are microorganisms that spends all or part of their life cycle within tissues of the plant host.1 A great deal of attention has been paid to fungal endophytes, an important component of plant-associated microbes, because of their functional diversity.2 Furthermore, endophytic fungi have been reported to be potential sources of compounds with intriguing chemical structures and biological activities, some of which are hopeful candidates for drug development.3, 4, 5 There have been reports of endophytes having the ability to protect host plants by producing secondary metabolites with biological activities including antibacterial, antifungal, antiviral, insecticidal, plant growth and regulatory activities. Moreover, endophytes may eventually help enhance the growth and competitiveness of host plants in nature. However, the interactions between them are still being investigated.1 Halophytes are plants that grow in highly saline waters, which is an environmental stressor of plants. Indeed, only 2% of all plant species are known to be halophytes. One of the mechanisms for halophytes to overcome salt stress is via an association between the halophyte and their endophytes.6 Even though endophytes were believed to help halophytes survive under saline conditions, only a few studies of secondary metabolites from the endophytes have been conducted.7 Herein, we attempted to elucidate bioactive secondary metabolites produced by endophytes isolated from halophytes in Suncheon Bay, which is an ecologically biodiverse area in South Korea. We isolated several endophytic fungi from a rhizome of a halophyte, Phragmites communis Trinius, collected from Suncheon Bay. In this study, Gaeumannomyces sp. JS0464 was selected for chemical investigation. Fungi belonging to the Harpophora-Gaeumannomyces species complex comprise one of the dark septate endophytes subgroups, which mainly colonize the roots of gramineous plant species.8, 9 Although plant pathogenic species are in the genus Gaeumannomyces (sexual stage of genus Harpophora), beneficial strains such as Harpophora oryzae are also included. Two strains (R-5-6 and RC-3-1) of H. oryzae isolated from the roots of wild rice (Oryza granulata) in China showed growth promoting activities on both wild and cultivated rice (O. sativa), and induced local and systemic resistance against rice blast caused by one of its close relatives, Magnaporthe oryzae.10 However, chemical components playing these beneficial roles have yet to be characterized.
In this study, secondary metabolites were isolated and identified from cultures of Gaeumannomyces sp. JS0464. Since dried rhizome of Phragmites communis is known to have neuroprotective effects and attenuate cognitive deficits,11 the anti-inflammatory activities of the isolated compounds were evaluated using a microglial cell assay.
Results and discussion
Among endophytic fungal strains isolated from the rhizome of P. communis, Gaeumannomyces sp. JS0464, was chosen for thorough chemical investigation because it showed a plethora of secondary metabolites during initial screening. The ethyl acetate soluble portion was subjected to repeated column chromatography over silica gel, C-18 and sephadex LH-20, followed by semi-preparative reverse-phase HPLC, leading to the isolation of two new glycosylated dialkylresorcinol derivatives (1–2), one new anthraquinone (3) and eight known compounds (4–11; Figure 1).

Structures of compounds 1–11 isolated from Gaeumannomyces sp.
Compound 1 was obtained as yellow needle crystals and its molecular formula was determined to be C21H34O6 by (+) high-resolution ESI-MS (HR-ESI-MS; m/z, 405.2249 [M+Na]+, calculated for C21H34O6Na, 405.2248). The 1H-NMR spectrum of 1 exhibited one aromatic signal at δ 6.15 (1H, s), seven methylene proton signals at δ 2.84 (1H, m), 2.56 (2H, m), 2.28 (1H, m), 1.54 (2H, m), 1.47 (2H, m), 1.36 (4H, m) and 1.35 (2H, m), and two methyl protons at δ 0.92 (3H, m) and 0.91 (3H, m). Six proton signals for a sugar moiety also appeared at δ 5.14 (1H, d, J=10 Hz), 4.23 (1H, d, J=3.6, 10 Hz), 4.09 (1H, q, J=7.2 Hz), 4.03 (1H, t, J=3.6 Hz), 3.82 (1H, d, J=3.6 Hz) and 1.51 (3H, d, J=7.2 Hz). The 13C-NMR spectrum showed 21 carbon signals corresponding to six aromatic carbons at δ 108.8, 114.7, 116.1, 141.4, 156.4 and 157.0, seven methylene carbons at δ 23.7, 23.7, 24.0, 32.3, 32.6, 33.0 and 34.6, two methyl signals at δ 14.4 and 14.5, and six carbons attributed to the sugar moiety at δ 16.2, 67.3, 69.0, 72.6, 73.3 and 76.3. Extensive analysis of one-dimensional- (1D) and 2D-NMR spectral data and comparison with those in the literature suggested that the aglycone unit was stemphol (4).12 HMBC correlations of the anomeric proton at δ 5.14 with C-3 (δ 157.0), C-4 (δ 114.7) and C-5 (δ 141.4) suggested that the sugar moiety was attached to C-4, as shown in Figure 2. The presence of a secondary methyl group together with five oxygenated carbons suggested that 1 had a 6-deoxyhexose, which was assigned as 6-deoxygulopyranose based on 1H–1H COSY correlations, 13C carbon chemical shifts and coupling constants (3J) between protons of the deoxyhexose. The large coupling constant for JH-1′′′,H-2′′′ (10 Hz) indicated a β-glycosidic linkage as well as H-2′′′ of the 6-deoxyhexose moiety in the axial position, suggesting that the hydroxyl group at C-2′′′ was in α orientation. Small coupling constants between H-2′′′ and H-3′′′ (3.6 Hz) suggested that H-3′′′ was equatorial since H-2′′′ was axial, indicating that the hydroxyl group at C-3′′′ was in α orientation. The broad quartet signal for H-5′′′ (J=7.2 Hz) along with small coupling constants for H-3′′′ and H-4′′′ suggested that the hydroxyl group was in β orientation. Based on these coupling constants of the proton signals in sugar moiety as well as comparison of the NMR resonances those in the literature, the deoxyhexose was elucidated as deoxygulose.13 Owing to the C-glycosidic linkage, compound 1 cannot be hydrolyzed, which made elucidation of absolute stereochemistry impossible by proper derivatization of the sugar moiety. Therefore, the structure of compound 1 was elucidated as resorcinol 2-butyl-5-pentyl-4-C-6-deoxy-β-gulopyranoside, and named stemphol C.

Observed 1H–1H COSY and key HMBC correlations of compounds 1 and 2.
Compound 2 was obtained as yellow amorphous powders. Its molecular formula was determined to be C21H34O6 by (+) HR-ESI-MS (m/z 405.2249 [M+Na]+, calculated for C21H20O9Na, 405.2248). 1D- and 2D-NMR spectra of 2 were quite similar to those of 1, differing in the chemical shifts and coupling constants of a 6-deoxyhexose moiety. The anomeric proton signal at δ 4.76 exhibited HMBC correlations with C-3 (δ 157.2), C-4 (δ 113.8) and C-5 (δ 139.2), indicating that the 6-deoxyhexose of 2 was also attached to C-4, as for compound 1. The broad singlet signal for the anomeric proton indicated an α-glycosidic linkage. The 6-deoxyhexose was determined to be α-rhamnose on the basis of 1H–1H COSY correlations and interpretation of coupling constants for JH-2,H-3 (2.9 Hz), JH-3,H-4 (9.3 Hz), JH-4,H-5 (9.0 Hz) and JH-1,H-2 (nearly 0 Hz).13 Thus, compound 2 was elucidated as resorcinol 2-butyl-5-pentyl-4-C-α-rhamnoside, and named stemphol D. The complete assignment of all proton and carbon atoms in compounds 1 and 2 is shown in Table 1.
Compound 3 was obtained as yellow amorphous powders, and its molecular formula was determined to be C21H20O9 by HR-ESI-MS (m/z 439.1001 [M+Na]+, calculated for C21H20O9Na, 439.1000). The 1H-NMR spectrum of 3 exhibited four aromatic signals at δ 7.84 (1H, br s), 7.72 (1H, d, J=2.6 Hz), 7.19 (1H, br s) and 7.17 (1H, d, J=2.4 Hz). Two proton signals at δ 3.92 (3H, s) and 2.30 (3H, s) were assigned to one methoxyl group directly attached to the aromatic carbons and one methyl proton. In addition, one anomeric proton signal appeared at δ 6.20 (1H, d, J=4.4 Hz). The 13C-NMR spectrum showed 21 carbon signals corresponding to two ketones (δ 187.9 and 183.2), 12 aromatic carbons (δ 108.7, 110.8, 113.0, 119.2, 119.9, 121.3, 135.1, 136.0, 147.6, 161.8, 164.2 and 165.6), 1 methoxy carbon (δ 56.9), 1 methyl carbon (δ 22.2) and 5 carbons for a pentose moiety (δ 63.3, 71.3, 74.2. 89.3 and 102.1). The 1H-NMR and 13C-NMR spectra (Table 2) indicated that 3 has an anthraquinone skeleton with a pentose. Extensive analysis of 1D- and 2D-NMR spectral data and comparison with those available in the literature suggested that the aglycone unit was 1-O-methylemodin (5). HMBC correlation of the methoxyl proton at δ 3.92 with C-1 (δ 161.8) indicated that the pentose moiety was attached to C-6 in anthraquinone. HMBC correlation of the methyl proton signal at δ 2.30 with C-2 (δ 119.9), C-3 (δ 147.6) and C-4 (δ 121.3) suggested that the methyl group was attached to C-3 (Figure 3). HMBC correlation of the anomeric proton (δH 6.20) with C-6 (δC 164.2) indicated that the pentose was attached to C-6 of the 1-O-methylemodin. The small coupling constant of anomeric proton (4.4 Hz) indicated an α-glycosidic linkage. The pentose was assigned as α-D-ribofuranose by comparison of the spectral data with those in the literature.14 Thus, the structure of compound 3 was elucidated as 1-O-methyl-6-O-(α-D-ribofuranosyl)-emodin.

Observed 1H–1H COSY (—) and key HMBC (→) correlations of compound 3.
The known compounds were identified as stemphol (4),12 1-O-methylemodin (5),15 2-(4-hydroxyphenyl)acetic acid (6),16 cycloartenyl ferulate (7),17 5α,8α-epidioxyergosta-6,9(11),22-trien-3-ol (8),18 ergosterol peroxide (9),19 5α,8α-epidioxy-(22E,24 R)-23-methylergosta-6,22-dien-3β-ol (10)20 and β-sitosterol (11) (Figure 1),21 respectively, by comparison of the NMR data with that in the literature.
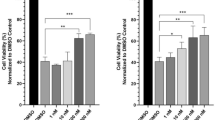
Anti-inflammatory activity of each compound was tested on microglia, a pivotal cell type implicated in neurodegeneration.22 With the exception of compounds 6 and 7, all compounds significantly reduced nitric oxide (NO) production in lipopolysaccharide (LPS)-stimulated BV-2 microglia cells, seven of which did not impair cell viability (Figure 4).

Effects of compounds from Gaeumannomyces sp. JS0464 on LPS-induced NO production and cell viability. (a) Effects of compounds on LPS-induced NO production. Microglial BV-2 cells pretreated with 50 μM compounds for 3 h were stimulated with LPS (1 μg ml–1) for 24 h. Griess reagent was used to detect the generation of extracellular nitrite at 24 h after the LPS challenge. (b) Effects of compounds on cell viability. BV-2 cells were treated with 50 μM compounds for 24 h before viability was measured. Data obtained from four biological replicates are shown as the means±s.e. values. Asterisks indicate significant differences identified by the Student's t-test (*P<0.05; **P<0.01; ***P<0.001). Abbreviations: LPS, lipopolysaccharide; NO, nitric oxide.
In particular, three new compounds (1–3) showed anti-inflammatory effects without causing cell death. Cytotoxicity caused by stemphol (4), a potent mycotoxin, was compromised by its derivatives (1 and 2). Moreover, an even increase in cell viability was observed in response to treatment with 2 (P<0.001). 1-O-methylemodin (5) and its new derivative (3) reduced NO production by LPS-treated cell by 43 and 31%, respectively, without causing cell death. 1-O-methylemodin (5) was first isolated from Basidiomycotina23 and shown to inhibit the growth of the tree decaying fungus Phellinus tremulae,24 the secretion of IL-625 and protein tyrosine phosphatase 1B.26 However, its anti-inflammatory activity has not yet been reported. Three ergosterol derivatives (8, 9 and 10) also abated LPS-induced NO levels. Compounds 8 and 10 did not hurt cell viability, while compound 9 showed cytotoxicity. Ergosterol is the main endogenous sterol produced by fungi and is known as one of active constituents of medicinal or edible mushrooms with diverse bioactivity including anti-cancer, immune-modulatory and anti-inflammatory activity.27 β-sitosterol (11), a representative plant sterol that is also found in rice bran,28 reduced NO production to 35% without cytotoxicity. β-sitosterol (11) has been shown to have anti-inflammatory activity in several cell models.29 Cycloartenyl ferulate (7), a derivative of rice bran oil-derived γ-oryzanol,30 has also been reported to have anti-inflammatory and anti-cancer activities, inducing apotosis.31 Thus, compounds 7 and 11 appeared to be derived from the rice media used in the culture.
In conclusion, a dark septate endophyte, Gaeumannomyces sp. JS0464, was isolated from a halophyte Phragmites communis and its chemical investigation led to the isolation of 11 secondary metabolites including 2 new C-glycosylated dialkylresorcinol derivatives (1–2) and a new glycosylated anthraquinone (3). Seven compounds, including three new compounds, showed anti-inflammatory activities against LPS-stimulated BV-2 microglia cells without cytotoxicity. The results presented herein suggest that endophytes from halophytes could be good resources for anti-inflammatory agents.
Experimental procedure
General experimental procedures
Optical rotation was measured using JASCO P-2000 polarimeter (Tokyo, Japan). NMR spectra were taken in DMSO-d6, CD3OD, and pyridine-d5, and chemical shifts were referenced relative to the corresponding signals (δH 3.31/ δC 49.15 for CD3OD; δH 8.74/ δC 150.35 for pyridine-d5) and measured on a Varian VNS 600 spectrometer (1H: 600 MHz, 13C: 150 MHz). The HR-ESI-MS were obtained using a Bruker UHR ESI Q-TOF mass spectrometer (Fremont, CA, USA). All HPLC were performed using Agilent series 1260 HPLC system (Palo Alto, CA, USA). Open column chromatography was carried out over a silica gel 60 (70–230 mesh, Merck, Germany), a LiChroprep RP-18 gel (40–63 um, Merck, Germany) and a Sephadex LH-20 gel (GE Healthecare, Sweden).
Isolation and cultures of a fungal strain
The fungal strain (JS0464) was isolated from the reed plant (Phragmites communis Trinius) collected from a swamp in Suncheon, South Korea (34° 83′ 79′′ N, 127° 44′ 95′′ E) in September 2011. Rhizome tissues were cut into small pieces (0.5 × 0.5 cm) and sterilized with 2% sodium hypochlorite for 1 min and 70% ethanol for 1 min, and then washed with sterilized distilled water. Fungal strains were cultured from plant tissues after about 7 days of incubation on malt extract agar (MEA, Difco, Sparks, MD, USA) amended with 50 p.p.m. kanamycin, 50 p.p.m. chloramphenicol and 50 p.p.m. Rose Bengal at 22 °C. Fungal strains were put into pure culture by transferring actively growing colony edges to a fresh potato dextrose agar (PDA, Difco) before being stored as 20% glycerol stocks in a liquid nitrogen tank at the Wildlife Genetic Resources Bank at the National Institute of Biological Resources (Incheon, Korea) before use. Fungal identification was conducted by sequencing internal transcribed spacer (ITS) regions with ITS1 and ITS4 primers.32 Phylogenetic analysis based on comparison of the ITS sequences with those from the NCBI resulted in JS0464 being grouped into a Gaeumannomyces (sexual stage)–Harpophora (asexual stage) species complex (Supplementary Figure 2). Since the phenotypic characteristics were different from those of Harpophora sp., in which thin colonies, phial-like conidiophores or sickle-shaped conidia were not observed, JS0464 was tentatively identified as Gaeumannomyces sp. The fungus was then cultured at 28 °C in still for 4 weeks in 20 × 500 ml Erlenmeyer flasks containing 80 g of rice and 120 ml of distilled water.
Extraction and isolation of metabolites
The fungal cultures grown on rice media were extracted with EtOAc (200 ml of each Erlenmeyer flasks) three times in a ultrasonic sonomatic cleaning bath (Branson 5510E-DTH, Danbury, CT, USA) at a frequency of 40 kHz for 1 h. The mixture solutions were then evaporated under reduced pressure at 45 °C to give EtOAc extracts (72.3 g). Next, the EtOAc extracts (70.0 g) were chromatographed with silica gel column chromatography using n-Hexane–EtOAc–MeOH gradient (v/v/v, 50:1:0→2:1:0.1→0:0:1) solvents to yield six fractions (A–F). Fraction C was separated by silica gel column chromatography using a gradient system of n-Hexane–EtOAc (v/v, 100:1→7:1) to achieve compound 8 (4.0 mg) and obtain 15 fractions (C1–C15). Fraction C9 was purified by HPLC with a C-18 column using a gradient system of H2O-MeOH (v/v, 95:5→0:100) to achieve compound 4 (18.0 mg). Fraction C14 was separated by silica gel column chromatography using a gradient system of n-Hexane–CH2Cl2–EtOAc (v/v/v, 5.5:1:0.1→1:1:0.1) to achieve compound 11 (8.0 mg). Fraction E was separated by silica gel column chromatography using a gradient system of n-Hexane–EtOAc–MeOH (v/v, 8:1:0→2:1:0.1) to yield 10 fractions (E1–12). Fraction E7 was subjected to C-18 column chromatography using an acetone–MeOH–water (v/v/v, 0:1:1→7:7:1) gradient system to obtain compound 7 (7.0 mg), 9 (5.7 mg) and 10 (4.7 mg). Fraction E8 was separated via crystallization to obtain compound 5 (2.8 mg). Fraction 11 was purified by HPLC with a C-18 column using a gradient system of H2O–MeOH (95:5→0:100) to achieve compound 1 (38.4 mg). Fraction 12 was subjected to C-18 column chromatography using a MeOH–water (1:1→4:1) gradient system to obtain compound 2 (6.2 mg) and 6 (9.9 mg). Fraction F was subjected to Sephadex LH-20 chromatography using 50% isocratic methanol to yield 20 fractions (F1-20). Fraction F9 was purified by HPLC with a C-18 column using a gradient solvent system of H2O–MeOH (v/v, 95:5→0:100) to achieve compound 6 (11.5 mg). Fraction F14 was purified by HPLC with a C-18 column using a gradient solvent system of H2O–MeOH (v/v, 95:5→0:100) to obtain compound 3 (1.8 mg).
Stemphol C: resorcinol 2-butyl-5-pentyl-4-C-6-deoxy-β-D-gulopyranoside (1): light yellow needle (MeOH/H2O); [α]D25=–23.4° (c 0.1, MeOH); 1H- and 13C-NMR data (Table 1); (+) HR-ESI-MS m/z, 405.2249 [M+Na]+, calculated for C21H34O6Na, 405.2248; 1H- and 13C-NMR (600 and 150 MHZ, CD3OD) spectroscopic analysis (Table 1). HMBC correlations (CD3OD, H-#→C-#) H-6→C-1, C-2, C-4, and C-5; H-1′→C-1, C-2. C-3, C-2′, and C-3′; H-1′′′→C-3, C-4, C-5, C-2′′′, and C-3′′′; H-1′′→C-4, C-5, C-6, C-2′′, and C-3′′; 1H–1H COSY correlations (CD3OD, H-#↔H-#) H-1′↔H-2′, H-2′↔H-3′, H-3′↔H-4′, H-1′′a↔H-1′′b and H-2′′, H-1′′b↔H-2′′, H-2′′↔H-3′′, H-3′′↔H-4′′, H-4′′↔H-5′′, H-1′′′↔H-2′′′, H-2′′′↔H-3′′′, H-3′′′↔H-4′′′, H-4′′′↔H-5′′′, H-5′′′↔H-6′′′.
Stemphol D: resorcinol 2-butyl-5-pentyl-4-C-α-L-rhamnoside (2): yellow amorphous powders (MeOH/H2O); [α]D25=+11.4 ° (c 0.1, MeOH); 1H- and 13C-NMR data (Table 1); (+) HR-ESI-MS m/z, 405.2249 [M+Na]+, calculated for C21H34O6Na, 405.2248; 1H- and 13C-NMR (600 and 150 MHZ, CD3OD) spectroscopic analysis, see Table 1. HMBC correlations (CD3OD, H-#→C-#) H-6→C-1, C-2, C-4, and C-5; H-1′→C-1, C-2. C-3, C-2′, and C-3′; H-1′′′→C-3, C-4, C-5, C-2′′′, and C-3′′'; H-1′′→C-4, C-5, C-6, C-2′′, and C-3′′; 1H–1H COSY correlations (CD3OD, H-#↔H-#) H-1′↔H-2′, H-2′↔H-3′, H-3′↔H-4′, H-1′′↔H-2′′, H-2′′↔H-3′′, H-3′′↔H-4′′, H-4′′↔H-5′′, H-1′′′↔H-2′′′, H-2′′′↔H-3′′′, H-3′′′↔H-4′′′, H-4′′′↔H-5′′′, H-5′′′↔H-6′′′.
1-O-methyl-6-O-(α-D-ribofuranosyl)-emodin (3): yellow amorphous powders; [α]D25=–42.3° (c 0.1, MeOH); 1H- and 13C-NMR data (Table 2); (+)HR-ESI-MS m/z 439.1001 [M+Na]+, calculated for C21H20O9Na, 439.1000; 1H- and 13C-NMR (600 and 150 MHZ, pyridine-d5) spectroscopic analysis, (Table 2). HMBC correlations (pyridine-d5, H-#→C-#) H-2→C-1, C-3, 3-CH3, C-4, and C-9a; H-4→C-2, C-3, 3-CH3, C-4a, C-9a and C-10′; H-5→C-6, C-7, C-8a, C-10 and C-10a; H-7→C-5, C-7, C-8 and C-8a; 3-CH3→C-2, C-3 and C-4, H-1′→C-6, C-2′, C-3′ and C-4′, 1-OCH3→C-1; 1H–1H COSY correlations (pyridine, H-#↔H-#) H-1′↔H-2′, H-2′↔H-3′, H-3′↔H-4′, H-4′↔H-5′a and H-5′b, H-5′a↔H-5′b.
No reduction assay
Murine microglial cells (BV-2), which were kindly provided by Dr Myung Sook Oh (Kyung Hee University, Korea), were grown in DMEM-low glucose (pH 7.2–7.4) media containing 4 g l–1 glucose, 3.7 g l–1 sodium bicarbonate, 10% fetal bovine serum and antibiotics (100 units ml–1 penicillin and 100 μg ml–1 streptomycin) in a humidified atmosphere with 5% CO2 at 37 °C. Overnight grown cells in 96-well clear bottom plates (Costar, Cambridge, MA, USA) at a concentration of 5 × 104 cells per well were treated with 1 μg ml–1 LPS for 24 h either with or without 3 h pre-treatment by compounds that were dissolved in DMEM medium, including 0.5% DMSO. For NO detection, cell culture supernatants were subjected to a colorimetric reaction with Griess reagent. Briefly, 50 μl of cell culture supernatants obtained at 24 h after LPS treatment were mixed with an equal (1:1) volume of Griess reagent. The absorbance was then measured at 540 nm using a 96-well microplate reader (Synergy HT, Bio-Tek Instruments, Inc., Winooski, VT, USA). Net absorbance of the product of the Griess reaction was obtained by subtracting the absorbance of the medium. Cell viability was evaluated using an MTS assay kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions. The quantity of formazan was measured at 490 nm, which is directly proportional to the mitochondrial dehydrogenase activity. A Student's t-test was conducted using the software R i386 3.3.0. Results are expressed as the means±s.e.
References
Tan, R. X. & Zou, W. X. Endophytes: a rich source of functional metabolites. Nat. Prod. Rep. 18, 448–459 (2001).
Su, Z. Z. et al. Evidence for biotrophic lifestyle and biocontrol potential of dark septate endophyte harpophora oryzae to rice blast disease. PLoS ONE 8, e61332 (2013).
Pudhom, K. & Teerawatananond, T. Rhytidenones A–F, Spirobisnaphthalenes from Rhytidhysteron sp. AS21B, an endophytic fungus. J. Nat. Prod. 77, 1962–1966 (2014).
Strobel, G. A. et al. Oocydin A, a chlorinated macrocyclic lactone with potent anti-oomycete activity from Serratia marcescens. Microbiology 145, 3557–3564 (1999).
Castillo, U. F. et al. Munumbicins, wide-spectrum antibiotics produced by Streptomyces NRRL 30562, endophytic on Kennedia nigriscans. Microbiology 148, 2675–2685 (2002).
Sgroy, V., Cassan, F., Masciarelli, O., Del Papa, M. F. & Luna, V. Isolation and characterization of endophytic plant-growth promoting or stress homeostasis-regulating bacteria associated to the halophyte Prosopis strombulifera. Appl. Microbiol. Biotechnol. 85, 371–381 (2009).
Suryanarayanan, T. S. & Kumaresan, V. Endophytic fungi of some halophytes from an estuarine mangrove forest. Mycol. Res. 104, 1465–1467 (2000).
Yuan, Z. L., Lin, F. C., Zhang, C. L. & Kubicek, C. P. A new species of Harpophora (Magnaporthaceae) recovered from healthy wild rice (Oryza granulata roots, representing a novel member of a beneficial dark septate endophyte. Fems. Microbiol. Lett. 307, 94–101 (2010).
Sieber, T. N. In Plant Roots: The Hidden Half 3rd edn (eds Waisel Y., Eshel A. & Kafkafi U.) Ch. 49, pp 887–917 (Marcel Dekker, 2002)..
Yuan, Z. L., Zhang, C. L., Lin, F. C. & Kubicek, C. P. Identity, diversity, and molecular phylogeny of the endophytic mycobiota in the roots of rare wild rice (Oryza granulate from a nature reserve in Yunnan, China. Appl. Environ. Microbiol. 76, 1642–1652 (2010).
Choi, J. H., Kim, I. S., Kim, J. I., Kim, D. W. & Yoon, T. H. Effect of reed root (Phraamites communis extract on physiological activity of rats. Kor. J. Gerontol. 3, 109–115 (1993).
Stodola, F. H., Weisleder, D. & Vesonder, R. F. A new dialkylresorcinol from Stemphylium majusculum. Phytochemistry 12, 1797–1798 (1973).
Liu, Q. et al. Antiproliferative cardiac glycosides from the latex of Antiaris toxicaria. J. Nat. Prod. 76, 1771–1780 (2013).
Li, D. L., Li, X. M. & Wang, B. G. Natural anthraquinone derivatives from a marine mangrove plant-derived endophytic fungus Eurotium rubrum: structural elucidation and DPPH radical scavenging activity. J. Microbiol. Biotechnol. 19, 675–680 (2009).
Fujimoto, H., Nakamura, E., Okuyama, E. & Ishibashi, M. Six immunosuppressive features from an ascomycete, Zopfiella longicaudata, found in a screening study monitored by immunomodulatory activity. Chem. Pharm. Bull. 52, 1005–1008 (2004).
Ohtani, K., Fujioka, S., Kawano, T., Shimada, A. & Kimura, Y. Z. Nematicidal activities of 4-hydroxyphenylacetic acid and oidiolactone D produced by the fungus Oidiodendron sp. Z. Naturforsch. C 66, 31–34 (2001).
Wang, W., Guo, J., Zhang, J., Peng, J., Liu, T. & Xin, Z. Isolation, identification and antioxidant activity of bound phenolic compounds present in rice bran. Food Chem. 171, 40–49 (2015).
Fangkrathok, N., Sripanidkulchai, B., Umehara, K. & Noguchi, H. Bioactive ergostanoids and a new polyhydroxyoctane from Lentinus polychrous mycelia and their inhibitory effects on E2-enhanced cell proliferation of T47D cells. Nat. Prod. Res. 27, 1611–1619 (2013).
Lee, I. S. et al. Cytotoxicity of ergosterol derivatives from the Fruiting Bodies of Hygrophorus russula. Nat. Prod. Sci 17, 85–89 (2011).
Yaoita, Y. et al. Sterol constituents from five edible mushrooms. Chem. Pharm. Bull. 46, 944–950 (1998).
Yu, B. C., Yang, M. C., Lee, K. H., Kim, K. H. & Lee, K. R. Norsesquiterpene and steroid constituents of Humulus japonicus. Nat. Prod. Sci. 13, 332–336 (2007).
Liu, B. & Hong, J. S. Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J. Pharmacol. Exp. Ther. 304, 1–7 (2003).
Ayer, W. A. & Trifonov, L. S. Anthraquinones and a 10-Hydroxyanthrone from Phialophora alba. J. Nat. Prod. 57, 317–319 (1994).
Gill, M. & Morgan, P. M. New fungal anthraquinones. Arkivoc 7, 145–156 (2001).
Yang, Y. et al. Anthraquinone derivatives from Rumex plants and endophytic Aspergillus fumigatus and their effects on diabetic nephropathy. Bioorg. Med. Chem. Lett. 23, 3905–3099 (2013).
Na, M. K., Jin, W. Y., Min, B. S., Ahn, J. S. & Bae, K. Protein tyrosine phosphatase 1B inhibitory activity of anthraquinones and stilbenes. Nat. Prod. Sci. 14, 143–146 (2008).
Hong, Y. J., Jang, A. R., Jang, H. J. & Yang, K. S. Inhibition of nitric oxide production, iNOS and COX-2 expression of ergosterol derivatives from Phellinus pini. Nat. Prod. Sci. 19, 147–152 (2012).
Sugano, M. & Tsuji, E. Rice bran oil and cholesterol metabolism. J. Nutr. 127, 521S–524S (1997).
Loizou, S., Lekakis, I., Chrousos, G. P. & Moutsatsou, P. Beta-sitosterol exhibits anti-inflammatory activity in human aortic endothelial cells. Mol. Nutr. Food Res. 54, 551–558 (2010).
Oka, T. et al. Cycloartenyl ferulate, a component of rice bran oil-derived gamma-oryzanol, attenuates mast cell degranulation. Phytomedicine 17, 152–156 (2010).
Kong, C. K. L. et al. A rice bran polyphenol, cycloartenyl ferulate, elicits apoptosis in human colorectal adenocarcinoma SW480 and sensitizes metastatic SW620 cells to TRAIL-induced apoptosis. Biochem. Pharmacol. 77, 1487–1496 (2009).
White, T. J., Bruns, T. D., Lee, S. D. & Taylor, J. W. in PCR Protocols: A Guide to Methods and Applications (eds Innis M. A., Gelfand D. H., Sninsky J. J. & White T. J.) Ch. 38, 315–322 (Academic Press, 1990)..
Acknowledgements
This research was supported by the National Institute of Biological Resources (NIBR201626101), Korea Institute of Planning and Evaluation for Technology in Food, Agriculture, Forestry and Fisheries (IPET) through High Value-added Food Technology Development Program, funded by Ministry of Agriculture, Food and Rural Affairs (MAFRA)(116001-3), and also Basic Science Research Program through NRF funded by the Ministry of Education (NRF-2015R1D1A1A01057914).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article

Cite this article
Lee, C., Kim, S., Li, W. et al. Bioactive secondary metabolites produced by an endophytic fungus Gaeumannomyces sp. JS0464 from a maritime halophyte Phragmites communis. J Antibiot 70, 737–742 (2017). https://doi.org/10.1038/ja.2017.39
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2017.39
This article is cited by
-
Do all fungi have ancestors with endophytic lifestyles?
Fungal Diversity (2023)
-
Endophytic fungi: a potential source for drugs against central nervous system disorders
Brazilian Journal of Microbiology (2023)
-
Insights into the beneficial roles of dark septate endophytes in plants under challenging environment: resilience to biotic and abiotic stresses
World Journal of Microbiology and Biotechnology (2022)
-
Bioactive compounds from an endophytic fungi Nigrospora aurantiaca
Botanical Studies (2021)
-
Chemical Constituents of the Endophyte Penicillium sp. Isolated from Artemisia princeps
Chemistry of Natural Compounds (2020)
