
Abstract
In order to modify lincomycin at the C-6 and C-7 positions, we prepared target molecules, which have substituted pipecolinic acid at the 6-amino group and a para-substituted phenylthio group at the C-7 position, in application of palladium-catalyzed cross-coupling as a key reaction. As the result of structure-activity relationship (SAR) studies at the 6-position, analogs possessing 4′-cis-(cyclopropylmethyl)piperidine showed significantly strong antibacterial activities against Streptococcus pneumoniae and Streptococcus pyogenes with an erm gene. On the basis of SAR, we further synthesized novel analogs possessing 4′-cis-(cyclopropylmethyl)piperidine by transformation of a C-7 substituent. Consequently, novel derivatives possessing a para-heteroaromatic-phenylthio group at the C-7 position exhibited significantly strong activities against S. pneumoniae and S. pyogenes with an erm gene even when compared with those of telithromycin. Finally, in vivo efficacy of selected two derivatives was evaluated in a rat pulmonary infection model with resistant S. pneumoniae with erm + mef genes. One of them exhibited strong and constant in vivo efficacy in this model, and both compounds showed strong in vivo efficacy against resistant S. pneumoniae with a mef gene.
Similar content being viewed by others

Introduction
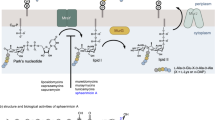
Macrolide antibiotics have antibacterial activities against S. pneumoniae, S. pyogenes, Haemophilus influenzae, Moraxella catarrhalis, Mycoplasma pneumoniae and Neisseria gonorrhoeae, and have an acceptable safety profile as oral antibiotics. Consequently, macrolides have widely been used in clinical sites for bacterial respiratory infections. Recently, macrolide resistant bacteria with an erm gene have markedly increased.1, 2, 3 Clarithromycin4 and azithromycin5, 6 are not effective against S. pneumoniae and S. pyogenes with an erm gene and have low sensitivity against S. pneumoniae with a mef gene (Figure 1 and Table 1). Although telithromycin (TEL)7 exhibits effective activities against S. pneumoniae with erm and/or mef genes, its activities are influenced by a mef gene. A serious liver damage8, 9 and loss of consciousness10, 11 were reported as side effects of TEL and medication with TEL was discontinued in Japan. Novel azalides reported by Miura et al.12, 13 are also effective against the above resistant pathogens, but these analogs are still under research process. Development of an oral antibiotic possessing potent antibacterial activities and an acceptable safety profile is strongly desired in clinical sites for respiratory infections.

Chemical structures of macrolide derivatives and lincomycin/clindamycin derivatives.
Lincomycin (LCM)14, 15, 16, 17 and clindamycin (CLDM)18 are effective against clinically isolated pathogens with a mef gene, but they are not effective against resistant bacteria with an erm gene (Figure 1, Table 1). As an overview, CLDM exhibits the following positive characters: (1) availability in p.o. and i.v. administrations (switch therapy is possible), (2) good distributions to the tissue and cells, (3) suppression19 of toxin production by streptococcal strains and (4) expected reasonable production cost of its derivatives compared with that of ketolides with a complex chemical structure. Thus, LCM derivatives might be clinically more valuable than ketolide antibiotics, if they are effective against Gram-positive pathogens with an erm gene.
Chemical modifications at the C-7 position of LCM were reported by several research groups.17, 18, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32 However, none of those compounds showed antibacterial activities against resistant Gram-positive pathogens with an erm gene. On the other hand, we reported that novel LCM derivatives modified at the C-7 position possessed antibacterial activities against resistant bacteria with an erm gene.33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43 In particular, compound 1 (Wakiyama et al.40) (Figure 1) and its analogues (possessing a ‘1-methylpiperidin-3-yl’ or ‘1-methyl-1,2,5,6-tetrahydropyridin-3-yl’ moiety instead of a ‘pyrimidin-5-yl’ moiety in compound 1) had significantly potent antibacterial activities against resistant bacteria with an erm gene.
Chemical modification at the C-6 position of LCM and/or CLDM was also performed by several research groups.17, 24, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54 The C-6 side chain, originally 1′-N-methyl-4′-trans-n-propylproline, has the following characters: (1) diastereoisomers with 2′,4′-trans configuration were more potent than cis isomers; (2) 1′-N-demethylclindamycin was twice as active in vitro against Sarcina lutea as CLDM, but 1′-N-demethyllincomycin was about one twentieth as active as LCM;21, 44, 45 (3) as for chain length (H, Me to octyl) at the 4′-position of LCM, the hexyl analog showed maximum in vitro antibacterial activity;21, 46 (4) introduction of hetero atoms to the 4′-side chain essentially lost activity;17, 21, 47 and (5) VIC-105403 (Lewis et al.24) (Figure 1) had potent activities compared with CLDM.
As another background information on chemical modifications at the C-6, azetidine,48, 49, 50, 51 piperidine48, 49, 52, 53, 54 and azepane analogs48, 49 were synthesized accompanied with modifications at the C-7. Regarding azetidine derivatives, 3′-trans-cyclobutylethyl CLDM derivative showed significant antibacterial activities against sensitive S. pneumoniae compared with CLDM, but 3′-trans-cyclopropylmethyl, 3′-trans-n-propyl and 3′-trans-n-butyl analogs exhibited similar potency as CLDM. As a piperidine derivative, 4′-cis-ethyl CLDM analog, pirlimycin, is used in mastitis therapy for cattle in the European countries and United States. On the other hand, VIC-105555 (Figure 1) was selected as a candidate, which exhibited preferable pharmacokinetics and characteristic in vitro antibacterial activities against methicillin-resistant Staphylococcus aureus (MRSA) and Enterococcus faecalis. Furthermore, azepane-type CLDM analogs were also synthesized and 5′-(3-fluorobutyl) analog was 32 times as active (MIC: 0.25 μg ml−1) against H. influenzae as CLDM (MIC: 8 μg ml−1).48
None of the C-6-modified compounds were disclosed to possess activities against Gram-positive-resistant bacteria with an erm gene. We reported that novel (7S)-substituted analogs42 modified at the N-1′ and C-4′ positions in a proline moiety had potent activities against Gram-positive-resistant bacteria with an erm gene. We further pursued modifications of LCM with a combination manner at the C-6 and C-7 positions in order to generate novel LCM derivatives exhibiting as strong antibacterial activities as TEL. Then, we synthesized novel (7S)-substituted analogs attached with piperidine or azepane instead of pyrrolidine (a part of proline) at the C-6 position. We have found three representative molecules so far and we chose a ‘pyrimidin-5-yl’-phenyl derivative (1) (Figure 1) as a C-7 side chain for optimization of a C-6 moiety, because a ‘1-methylpiperidin-3-yl’ moiety has a chiral center (anxiety for relatively complex production) and a ‘1-methyl-1,2,5,6-tetrahydropyridin-3-yl’ moiety has an isolated double bond (anxiety for potential instability).
Results and discussion
Synthesis of the substituted piperidines and 2,3,6,7-tetrahydro-1H-azepine
Syntheses of substituted piperidines and 2,3,6,7-tetrahydro-1H-azepine are shown in Scheme 1. Substituted piperidines (±)-5-7 and 2,3,6,7-tetrahydro-1H-azepine 16 were synthesized by methods reported by Shuman et al.55 and Lewis et al.48 Compound (±)-12 was prepared from 4-methylpyridine (8) in improved reaction conditions based on reported methods48, 55 shown in Scheme 1. It was reported that hydrogenation of disubstituted pyridine in the presence of PtO2 resulted in a racemate of cis-products as major products by Lewis et al.48 Going back in time, Shuman et al.55 proved that 2,4-disubstituted piperidine prepared from disubstituted pyridine by hydrogenation had cis-configuration by NOE experiments. At the beginning of this research, we used (±)-cis-carboxylic acids 5-7 and 12, but later on we could separate (±)-12 into each cis-enantiomer for efficient synthetic study. Carboxylic acid (±)-12 was protected by a benzyl group for the purpose of optical resolution by HPLC and both enantiomers were purified by chiral column chromatography to obtain a desired compound 13. Stereochemistry of compounds 13 and 14 was assigned as following. Pirlimycin and VIC-105555 are reported as representative LCM derivatives possessing a substituted piperidine moiety (Figure 1). Absolute stereochemistry of pirlimycin was clarified by X-ray crystallographic studies.49 Absolute stereochemistry of VIC-105555 was reported by Vicuron at 44th Interscience Conference on Antimicrobial Agents and Chemotherapy.54 Both compounds have 2′-β-4′-β-configuration and they showed remarkable polarity (lower Rf value) and stronger potency compared with the corresponding 2′-α-4′-α-diastereoisomer, respectively. When we coupled a substituted pipecolic acid with methyl α-thiolincosaminide (MTL), we assigned 2′-β-4′-β-configuration for a polar product. The benzyl group in 13 was removed by hydrogenolysis to give a key intermediate 15. A seven-membered intermediate (17) was prepared by basic hydrolysis of 16.
Synthesis of key intermediates 30–33 and 39
Syntheses of key intermediates 30–33 and 39 are shown in Scheme 2. Diastereomeric mixtures 18–21 and compound 34 were synthesized by coupling of compounds (±)-5-7, (±)-12 and 17 with MTL, respectively. MTL was prepared by a reported method.56 Although each isomer was almost one to one mixture except 17 when the coupling reactions were completed, precipitation process gave 2′-β-4′-β-rich cis-isomers. As ‘Experimental procedure’ reported, ratio of diastereoisomeric mixture was difference for each compound. Tetra-O-trimethylsilylation of mixtures 18–21 and regioselective deprotection of the 7-O-TMS group followed by silica gel column chromatography finally gave single compounds 22–2548, 57 as 2′-β-4′-β-pure cis-isomers. Methanesulfonylation of the 7-OH group and then SN2 reaction by potassium thioacetate gave compounds 26-29. Key intermediates 30-3333, 34, 35, 37, 38, 40, 42 were prepared by deprotections of all TMS groups and an acetyl group. On the other hand, the Ns group of compound 34 was deprotected by 4-bromobenzenethiol under the basic condition, and then the olefin group was reduced by hydrogenation to give an azepane intermediate 35 (stereochemistry at the C-5′ position is not assigned). An amino group of 35 was protected by a Boc group to give 36, and a key intermediate 39 was synthesized from 36 by the similar procedures as described for the preparation of 30.
Synthesis of novel (7S)-4-(pyrimidin-5-yl)phenylthio LCM derivatives possessing piperidine or azepane as the C-6 side chain
Syntheses of novel (7S)-4-(pyrimidin-5-yl)phenylthio LCM derivatives possessing piperidine or azepane as the C-6 side chain are shown in Scheme 3. Compounds 40-43 and 48 were synthesized from key intermediates 30-33 and 39 by palladium-catalyzed cross-coupling reaction with 5-(4-bromophenyl)pyrimidine, respectively.33, 34, 35, 38, 40, 42, 58 The Boc group of 40-43 and 48 was finally removed with TFA to give desired compounds 44-47 and 49.
Synthesis of divergent intermediate 54 and novel LCM derivatives possessing a 4-(2-(dimethylamino)ethyl)phenylthio group at the C-7 position
Because we had to develop a more divergent synthetic route than those exemplified in Schemes 2 and 3, we decided to apply the next key intermediate 54. Syntheses of divergent intermediate 54 and novel LCM derivatives possessing a 4-(2-(dimethylamino)ethyl)phenylthio group at the C-7 position are shown in Scheme 4. Compound 5059 was synthesized by trifluoroacetylation of an amino group of MTL, and tetra-O-trimethylsilylation of all OH groups of 50 gave compound 51. Divergent intermediate 54 was synthesized from 51 by the similar procedures as described for preparation of 30. Palladium-catalyzed cross-coupling reaction of 54 with 2-(4-bromophenyl)-N,N-dimethylethanamine gave 55, which was hydrolyzed in the presence of phase transfer catalyst under the basic condition to give diamine 56. A coupling reaction of 56 with enantio-pure 15 provided desired 57 with all carbon’s framework. Deprotection of the Boc group finally gave 58 and its reductive N-methylation provided compound 59.
Synthesis of novel 4′-cis-(cyclopropylmethyl)piperidine LCM derivatives possessing a 4-substituted phenylthio group at the C-7 position
Syntheses of novel 4′-cis-(cyclopropylmethyl)piperidine LCM derivatives possessing a 4-substituted phenylthio group at the C-7 position are shown in Scheme 5. Compounds 60, 61 and 63-65 were synthesized from the key intermediate 33 by palladium-catalyzed cross-coupling reaction with the corresponding 4-substituted phenyl bromides. Reduction of 61 afforded saturated N-methylpiperidine 62 as a mixture of diastereoisomers at an N-methylpiperidine ring. The first half of desired compounds 66, 68, 70, 72, 74 and 77 were prepared by deprotection of a Boc group and their free secondary amine was methylated by reductive alkylation to give the second half of desired compounds 67, 69, 71, 73, 75 and 78, respectively. Compound 76 was also synthesized from 47 with the similar procedure. We confirmed that compound 76 derived from compound 33 had 4′-cis-stereochemistry by ROESY experiments. As the above, 4′-cis-stereochemistry of compound 12 was assigned.
SAR analysis of C-6 modified and (7S)-7-(4-(pyrimidin-5-yl)phenyl)thio-substituted LCM derivatives 44-47 and 49
We reported potent antibacterial activities of 1 possessing the (7S)-(4-(pyrimidin-5-yl)phenyl)thio group at the C-7 position. For the purpose of generating novel compounds possessing more potent antibacterial activities against resistant Gram-positive pathogens with an erm gene, we performed an SAR analysis of C-6-modified and (7S)-7-(4-(pyrimidin-5-yl)phenyl)thio-substituted LCM derivatives 44–47 and 49 (Table 2). According to our reported SAR studies, (7S) stereochemistry was selected among all novel derivatives.40 Compound 44, which possesses n-propyl-piperidine instead of n-propyl-pyrrolidine as the C-6 side chain, showed stronger activities against resistant S. pneumoniae with an erm gene than 1. However, its antibacterial activity against resistant S. pneumoniae with both erm and mef genes (S. pneumoniae-6) was not sufficient (MIC: 1 μg ml−1). Because there were a couple of reports21, 42, 46 stating that elongation of a side chain in a piperidine ring enhanced antibacterial activity, we synthesized alternative derivatives with a longer carbon chain or a branched side chain. However, antibacterial activities of compounds 45 and 46 were not improved. On the other hand, both compounds 47 and 49 possessing a 4′-cis-(cyclopropylmethyl)piperidine-2-carbonyl and 5′-n-propylazepane-2-carbonyl group at the C-6 position exhibited potent antibacterial activities against resistant S. pneumoniae with erm gene. Because the cyclopropylmethyl analog (47) especially exhibited stronger activities against Gram-positive pathogens with an erm gene even compared with TEL, we chose a 4′-cis-cyclopropylmethyl moiety as the C-6 side chain for further medicinal chemistry.
Antibacterial activities of novel LCM derivatives 58, 59 and 66–71 possessing an aliphatic amine at the para-position of phenylthio group at the C-7 position
For the purpose of accumulating detail information of SAR on (7S)-7-(4-substituted-phenylthio) LCM derivatives with a 4′-cis-(cyclopropylmethyl)piperidine moiety, we synthesized novel derivatives possessing various substituents at the C-7 position with a set of R2=both ‘N-H’ and ‘N-Me’ analogs (Table 3). Consequently, compounds 58, 66 and 68–71 showed potent antibacterial activities against target pathogens with an erm gene and their activities were relatively stronger even when compared with those of TEL. In addition, antibacterial activity of all compounds against S. pyogenes with an erm gene was significantly potent than that of TEL. We confirmed that combination of chemical modifications with the 4′-cis-(cyclopropylmethyl)piperidine group at the C-6 position and an aliphatic amine to the para-position of a phenylthio group at the C-7 position was important to enhance antibacterial activities against S. pneumoniae and S. pyogenes with an erm gene.
Antibacterial activities of novel LCM derivatives 47 and 72–78 possessing an aromatic amine at the para-position of a phenylthio group at the C-7 position
In order to expand possibilities of the combination modification at both the C-6 and C-7 positions, we synthesized novel derivatives possessing various aromatic amines as a substituent on the phenyl group with a set of both ‘N-H’ and ‘N-Me’ analogs (Table 4). All their antibacterial activities against target Gram-positive pathogens with an erm gene were also relatively stronger than those of TEL. To be more precise, compounds 47 and 72 showed potent activities in ‘N-H’ analogs and compound 76 exhibited the strongest activities among all ‘N-Me’ analogs in this article. As pharmacokinetic property must be different between ‘N-H’ and ‘N-Me’ analogs, it is important to select these two types of analogs for further development. Furthermore, antimicrobial activity of compounds 72, 47 and 77 against H. influenzae was relatively strong among all LCM derivatives we reported, and their potency was stronger than that of clarithromycin and catching up with that of TEL. We also investigated antibacterial activity against M. pneumoniae (Table 4), because resistant M. pneumoniae is causing problems for respiratory infections in clinical sites. All evaluated compounds including 47 and 76 had significant antibacterial activity against resistant M. pneumoniae, which TEL was not effective against. We could generate several novel LCM derivatives exhibiting very strong antibacterial activities against resistant Gram-positive pathogens with erm and/or mef genes by combination modification at the C-6 position (the proline moiety) and the C-7 position. These derivatives were also effective against resistant M. pneumoniae.
In vitro antibacterial activity (sensitivity distribution analysis) of selected compounds against sixty clinical isolates of S. pneumoniae
We evaluated the antibacterial activity of compounds 47, 68, 72, 76, 77 and TEL against 60 clinical isolates of S. pneumoniae including susceptible strains and resistant strains with erm and/or mef genes for sensitivity distribution analysis (Figure 2). MIC90 values of five novel LCM derivatives (0.06–0.125 μg ml−1) were relatively smaller than that of TEL (0.25 μg ml−1). Notably, 47 and 72 were significantly potent among tested compounds. These results reflect MIC values in Table 4 and it was suggested that these derivatives would also be effective against S. pneumoniae in clinical sites.

In vitro antibacterial activity (sensitivity distribution) of compounds 47, 68, 72, 76, 77 and TEL against 60 clinical isolates of S. pneumoniae.
In vivo efficacy of 47 and 76 (subcutaneous administration) in rat pulmonary infection model with resistant S. pneumoniae with erm + mef genes and a mef gene
We finally investigated the in vivo efficacy of selected compounds in rat pulmonary infection model with resistant S. pneumoniae with erm + mef genes. Among derivatives reported in this study, in vitro activities of compounds 47 and 72 are rather strong (Figure 2). On the other hand, we had to clarify in vivo efficacy in the set of ‘N-H’ and ‘N-Me’ in the piperidine moiety (to evaluate ‘72 and 73’ or ‘47 and 76’). As in vitro activities of 73 were slightly weaker than those of 76, we decided to select the set of compounds 47 and 76 for in vivo evaluation. Compound 72 had weak hemolytic activity and thus compound 72 might not be appropriate for further evaluation. Compounds 47, 76 and TEL were subcutaneously administered (10 mg kg−1) to rats at 2 h after bacterial infection, and in vivo efficacies are shown in Figure 3a. Compound 47 exhibited strong in vivo efficacy (3 log reduction or more) against resistant S. pneumoniae with erm + mef genes and its efficacy was constant (small s.d. value) compared with that of TEL (<2 log reduction). For our references, we also evaluated in vivo efficacy of those by subcutaneous administration (3 mg kg−1) to rats at 2 h after bacterial infection with S. pneumoniae with a mef gene, because resistant strains with a mef gene have increased in the US.60 As a result shown in Figure 3b, 47 and 76 had significantly strong in vivo efficacy as expected on the basis of in vitro evaluation. In vivo efficacy of 76 (5 log reduction) was very constant (0 s.d. value) compared with that of TEL. Clinical efficacy of these novel LCM derivatives is expected from the above fundamental experimental data.

(a) In vivo efficacy of 47 and 76 in a rat pulmonary infection model with S. pneumoniae MSC06856 (erm + mef). (b) In vivo efficacy of 47 and 76 in a rat pulmonary infection model with S. pneumoniae MSC6729 (mef). Comparison of the efficacy of novel lincomycin derivatives 47 and 76 in a rat pulmonary neutropenic infection model with S. pneumoniae MSC06856 (erm + mef) and S. pneumoniae MSC06729 (mef). Three rats per group were rendered neutropenic and 106 CFU per rat of S. pneumoniae MSC06856 or S. pneumoniae MSC06729 was injected into the lung, followed by s.c. administration of the test compounds at 2 h after infection. The mean log10 CFU per lung recovered from the infected lung after 24 h is shown. Error bars represent the s.d.
Conclusion
As the result of SAR studies at the 6-position of 44–47 and 49, compound 47 possessing 4′-cis-(cyclopropylmethyl)piperidine showed significantly strong antibacterial activities against S. pneumoniae and S. pyogenes with an erm gene. On the basis of SAR, we synthesized novel analogs possessing 4′-cis-(cyclopropylmethyl)piperidine by transformation of a C-7 substituent. Consequently, compounds 47, 68, 72, 76 and 77 (Figure 4) exhibited significantly strong activities against S. pneumoniae and S. pyogenes with an erm gene even when compared with those of TEL. Then, the in vitro antibacterial activities of compounds 47, 68, 72, 76, 77 and TEL were evaluated (sensitivity distribution analysis) against 60 clinical isolates of S. pneumoniae containing sensitive bacteria and resistant bacteria with erm and/or mef genes. As a result, compounds 47 and 72 showed relatively strong activities than that of TEL. Finally, the in vivo efficacy of compound 47 and its 1′-N-Me-derivative 76 was evaluated in the rat pulmonary infection model (subcutaneous administration) with resistant S. pneumoniae with erm + mef genes. Compound 47 exhibited strong and constant in vivo efficacy. Moreover, compounds 47 and 76 showed strong in vivo efficacy against resistant S. pneumoniae with a mef gene. These two compounds are under consideration toward next developing stage.

Structures of novel lincomycin derivatives possessing strong in vitro antibacterial activity.
Experimental procedure
General methods
1H NMR spectra were measured with a BRUKER Ascend 400 NMR spectrometer (BRUKER Corporation, Coventry, UK) for 400 MHz, JEOL JNM-GSX 400 NMR spectrometer (JEOL Ltd, Tokyo, Japan) for 400 MHz or a Varian Gemini 300 NMR spectrometer (Varian Inc., Palo Alto, CA, USA) for 300 MHz in CDCl3 or CD3OD. TMS (0 p.p.m.) in CDCl3 or CD3OD was used as an internal reference standard. Mass spectra were obtained on a JEOL JMS-700 mass spectrometer (JEOL Ltd) or Agilent Technologies 6530-Q-TOF–LC/MS mass spectrometer (Agilent Technologies, Santa Clara, CA, USA). The optical rotations were recorded with Jasco P-2300 digital polarimeter (JASCO Corporation, Tokyo, Japan). Column chromatography was performed with silica gel (Wakogel C200, Wako Pure Chemical Industries Ltd, Osaka, Japan). Preparative TLC was performed with silica gel (Merck, Darmstadt, Germany: TLC plates Silica gel 60 F254). All organic extracts were dried over anhydrous MgSO4 and the solvent was removed with a rotary evaporator under reduced pressure.
4-(Cyclopropylmethyl)pyridine (9)
To a solution of 8 (19.0 g, 204 mmol) in THF (136 ml) at −78 °C was added 2.0 M lithium diisopropylamide in tetrahydrofuran (THF) solution (204 ml, 408 mmol) and then was stirred in argon atmosphere at −40 °C for 20 min. The mixture was cooled to −78 °C. Then, bromocyclopropane (16.3 ml, 204 mmol) was added with dropwise to the solution. After stirring for 1 h, the solution was poured into saturated aqueous NH4Cl. The desired compound was extracted with ethyl acetate, was washed with brine and then the organic phase was dried over Na2SO4, filtrated and concentrated under reduced pressure. The resulting residue was purified by distillation under reduced pressure (84 °C/8 mm Hg) to obtain the title compound (13.8 g, 51%) as colorless oil. Fast atom bombardment (FAB)–MS m/z 134 (M+H)+ as C9H11N; 1H NMR (400 MHz, CDCl3) δ 0.18–0.26 (m, 2 H), 0.54–0.62 (m, 2 H), 0.93–1.05 (m, 1 H), 2.54 (d, J=7.1 Hz, 2 H), 7.17–7.23 (m, 2 H), 8.47–8.53 (m, 2 H).
4-(Cyclopropylmethyl)picolinonitrile (10)
To a solution of 9 (25.5 g, 191 mmol) in CH2Cl2 (300 ml) at 0 °C was added m-chloroperoxybenzoic acid (50.8 g, 191 mmol) and stirred at room temperature for 1 h. To the mixture was added Na2S2O3 solution (75 g in 150 ml of H2O). The solution was added to mixture of saturated aqueous NaHCO3 (500 ml), saturated aqueous K2CO3 (40 ml) and CHCl3 (500 ml). The organic phase was separated and then further extracted twice with CHCl3 (500 ml)-isopropanol (100 ml), the organic phase was dried over Na2SO4, filtrated and concentrated under reduced pressure to obtain 4-(cyclopropylmethyl)pyridine N-oxide (30.7 g as crude). 1H NMR (400 MHz, CDCl3) δ 0.15–0.26 (m, 2 H), 0.54–0.66 (m, 2 H), 0.90–1.02 (m, 1 H), 2.54 (d, J=7.1 Hz, 2 H), 7.11–7.24 (m, 2 H), 8.08–8.18 (m, 2 H).
To a solution of 4-(cyclopropylmethyl)pyridine N-oxide (30.7 g) in CH2Cl2 (350 ml) were added trimethylsilanecarbonitrile (30.6 ml, 0.229 mmol) and dimethylcarbamic chloride (7.03 ml, 76.3 mmol) at room temperature. Then, dimethylcarbamic chloride (7.03 ml, 76.3 mmol) was added in two portions after 20 min interval to the mixture at 20 °C. The mixture was stirred at room temperature for 17 h. The solution was added to 10% aqueous K2CO3. The desired compound was extracted with CH2Cl2 and then the organic phase was dried over Na2SO4, filtrated and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane to hexane/ethyl acetate=17/3) to obtain the title compound (25.5 g, 84% in 2 steps) as a colorless oil. EI–MS m/z 158 (M)+ as C10H10N2; 1H NMR (400 MHz, CDCl3) δ 0.22–0.28 (m, 2 H), 0.60–0.70 (m, 2 H), 0.92–1.06 (m, 1 H), 2.61 (d, J=7.1 Hz, 2 H), 7.41–7.46 (m, 1 H), 7.64 (br dd, J=1.7, 0.7 Hz, 1 H), 8.55–8.64 (m, 1 H).
4-(Cyclopropylmethyl)picolinic acid (11)
To a solution of 10 (25.5 g, 161 mmol) in MeOH (300 ml) was added 5 M aqueous NaOH (250 ml) and stirred at 50 °C for 8 h. The mixture was cooled down to 0 °C, added to 5 M aqueous HCl (250 ml) at 0 °C and then concentrated under reduced pressure to remove MeOH. The solution was adjusted at pH 3 by 1 M aqueous HCl, extracted with CHCl3 (500 ml)-isopropanol (150 ml) and then the organic phase was dried over Na2SO4, filtrated and concentrated under reduced pressure to obtain the title compound (27.6 g, 97%) as a colorless solid. FAB–MS m/z 178 (M+H)+ as C10H11NO2; 1H NMR (400 MHz, CDCl3) δ 0.22-0.30 (m, 2 H), 0.60–0.67 (m, 2 H), 0.95–1.11 (m, 1 H), 2.68 (d, J=7.1 Hz, 2 H), 7.48–7.54 (m, 1 H), 8.15-8.20 (m, 1 H), 8.58 (d, J=5.1 Hz, 1 H).
N-(tert-Butoxycarbonyl)-4-(cyclopropylmethyl)piperidine-2-carboxylic acid ((±)-12)
To a solution of 11 (6.90 g, 38.9 mmol) in AcOH (62 ml) was added PtO2 (442 mg) and then vigorously stirred in hydrogen atmosphere at room temperature for 24 h. The mixture was filtrated with celite and concentrated under reduced pressure to obtain 4-(cyclopropylmethyl)piperidine-2-carboxylic acid (5.3 g as crude). For the qualified analytical purpose, the above crude compound was purified by reverse-phase column chromatography (0.1% aqueous TFA/CH3CN=90/10 to 10/90) to obtain the highly purified 4-(cyclopropylmethyl)piperidine-2-carboxylic acid TFA salt as a colorless solid. ESI–MS m/z 184 (M+H)+ as C10H17NO2; 1H NMR (400 MHz, D2O) δ −0.08 to 0.02 (m, 2 H), 0.31–0.41 (m, 2 H), 0.58–0.72 (m, 1 H), 1.10–1.43 (m, 4 H), 1.71–1.85 (m, 1 H), 1.92–2.03 (m, 1 H), 2.35–2.45 (m, 1 H), 2.91–3.03 (m, 1 H), 3.39–3.48 (m, 1 H), 3.85–3.94 (m, 1 H).
To a solution of the above disubstituted piperidine (5.3 g) in 1,4-dioxane (100 ml) were added 2 M aqueous NaOH (74.0 ml, 148 mmol) and di-tert-butyl dicarbonate (14.3 ml, 62.2 mmol) and then stirred at room temperature for 15 h. The mixture was concentrated under reduced pressure to remove 1,4-dioxane. The solution was adjusted at pH 8 by1 M aqueous NaOH. Then, to the aqueous phase was added H2O, washed with Et2O, adjusted at pH 4 by 1 M aqueous HCl, extracted with ethyl acetate and then the organic phase was dried over Na2SO4, filtrated and concentrated under reduced pressure to obtain the title compound (10.5 g, 96% in 2 steps) as a racemate of cis-isomers as a colorless solid. It was reported that hydrogenation of disubstituted pyridine in the presence of PtO2 resulted in an approximately 1:1 mixture of two isomeric cis-products by Birkenmeyer et al.49 1H NMR (400 MHz, CDCl3) δ −0.04 to 0.08 (m, 2 H), 0.36–0.50 (m, 2 H), 0.60–0.73 (m, 1 H), 1.12–1.32 (m, 2 H), 1.35–1.55 (m, 1 H), 1.45 (s, 9 H), 1.66–1.89 (m, 3 H), 2.02–2.15 (m, 1 H), 3.55–3.60 (m, 2 H), 4.22–4.35 (m, 1 H).
2-Benzyl (2S, 4R)-N-(tert-butyl)-4-(cyclopropylmethyl)piperidine-1,2-dicarboxylate (13) 2-Benzyl (2R, 4S)-N-(tert-butyl)-4-(cyclopropylmethyl)piperidine-1,2-dicarboxylate (14)
To a solution of (±)-12 (113 mg, 0.399 mmol) in CH3CN (1 ml) were added diisopropylethylamine (0.104 ml, 0.599 mmol) and benzylbromide (0.652 ml, 0.439 mmol), and then stirred at room temperature for 48 h. The solution was added to saturated aqueous NaHCO3. The desired compound was extracted with ethyl acetate and then the organic phase was dried over Na2SO4, filtrated and concentrated under reduced pressure. The resulting residue was purified by preparative TLC (hexane/ethyl acetate=5/1) to obtain 2-benzyl N-(tert-butyl) 4-(cyclopropylmethyl)piperidine-1,2-dicarboxylate (133 mg, 89.3%) as a colorless oil. The above colorless oil (32.0 g) was further purified by column chromatography (Chiralpak AD-H, n-hexane/IPA=98/2) to obtain 13 (11.1 g, 35%) and 14 (11.0 g, 34%) as a colorless solid both. These enantiomers could be independently analyzed by the following condition: Chiralpak AD-H, 0.46 cm I.D × 25 cm, n-hexane/IPA=98/2, 1.0 ml min−1, 40 ºC, 208 nm. 13: [α]D27 −24.8° (c 0.65, CHCl3); ESI–MS m/z 374 (M+H)+ as C22H31NO4; 1H NMR (400 MHz, CDCl3) δ −0.12 to −0.02 (m, 2 H), 0.32–0.44 (m, 2 H), 0.52–0.66 (m, 1 H), 0.99–1.18 (m, 2 H), 1.30–1.50 (m, 1 H), 1.41 (s, 9 H), 1.69–1.88 (m, 3 H), 2.04 (ddd, J=13.5, 6.5, 4.6 Hz, 1 H), 3.08–3.70 (m, 2 H), 4.38 (t, J=6.3 Hz, 1 H), 5.08–5.23 (m, 2 H), 7.27–7.42 (m, 5 H). 14: [α]D26 +24.2° (c 1.03, CHCl3); ESI–MS m/z 374 (M+H)+ as C22H31NO4. Compound 14 showed the exactly same 1H NMR spectrum with that of 13.
(2S, 4R)- N-(tert-Butoxycarbonyl)-4-(cyclopropylmethyl)piperidine-2-carboxylic acid (15)
To a solution of 13 (1.51 g, 4.04 mmol) in MeOH (45 ml) was added Pd/C (0.166 g) and then vigorously stirred in hydrogen atmosphere at room temperature for 1 h. The mixture was filtrated with celite and concentrated under reduced pressure to obtain the title compound (1.15 g, quant) as a colorless solid. [α]D27 −16.3° (c 0.40, MeOH); ESI–MS m/z 284 (M+H)+ as C15H25NO4; time-of-flight (TOF)–ESI–HR-MS (M−H)− calcd for C15H25NO4: 282.1705, found: 282.1718 ; Compound 15 showed the exactly same 1H NMR spectrum with that of (±)-12.
(2S, Z)-1-(2-Nitrophenylsulfonyl)-5-n-propyl-2, 3, 6, 7-tetrahydro-1H-azepine-2-carboxylic acid (17>)
To a solution of 16 (72 mg, 0.19 mmol) in 1,4-dioxane (0.8 ml)-H2O (0.2 ml) was added LiOH·H2O (23.7 mg, 0.56 mmol) and then stirred at room temperature for 5 h. The mixture was diluted with H2O, washed with Et2O. The aqueous phase was adjusted at pH 3 by citric acid. The desired compound was extracted with ethyl acetate and then the organic phase was dried over Na2SO4, filtrated and concentrated under reduced pressure to obtain the title compound (70 mg as crude). This crude compound including a trace amount of citric acid could not be purified. ESI–MS m/z 369 (M+H)+ as C16H20N2O6S; 1H NMR (400 MHz, CD3OD) δ 0.73 (t, J=7.4 Hz, 3 H), 1.16–1.31 (m, 2 H), 1.73–1.82 (m, 2 H), 2.15–2.26 (m, 2 H), 2.41–2.51 (m, 1 H), 2.62–2.70 (m, 1 H), 3.42 (ddd, J=14.5, 7.8, 5.2 Hz, 1 H), 3.72 (dt, J=14.6, 4.7 Hz, 1 H), 4.71 (dd, J=7.1, 3.7 Hz, 1 H), 5.37 (t, J=6.5 Hz, 1 H), 7.56–7.71 (m, 3 H), 7.95-8.03 (m, 1 H).
Mixture 18 of methyl 6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(n-propyl)piperidine-2′-carbonyl)-α-thiolincosaminide and methyl 6-N-((2′R, 4′S)-1′-N-(tert-butoxycarbonyl)-4′-(n-propyl)piperidine-2′-carbonyl)-α-thiolincosaminide
To a solution of (±)-5 (11.7 g, 43.1 mmol) in DMF (100 ml) were added 1-hydroxybenzotriazole (7.55 g, 55.8 mmol), N,N′-dicyclohexylcarbodiimide (10.7 g, 51.9 mmol) and MTL (14.2 g, 56.1 mmol) and stirred at room temperature for 12 h. To the mixture was added H2O and then the solution was filtrated, and ethyl acetate and saturated aqueous NaHCO3 were added to the filtrate. The desired compound was extracted with ethyl acetate, extracted with CHCl3 and then the organic phase was dried over Na2SO4, filtrated and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane/ethyl acetate=50/50 to ethyl acetate, then ethyl acetate to ethyl acetate/ MeOH=90/10) to obtain 6-N-(N′-(tert-butoxycarbonyl)-4′-(n-propyl)piperidine-2′-carbonyl)-α-thiolincosaminide (18.5 g, 84.9%, (2′S, 4′R) isomer:(2′R, 4′S) isomer=ca. 50:50) as a colorless solid. To this colorless solid was added ethyl acetate, and insoluble matter was filtrated off and ethyl acetate solution was concentrated under reduced pressure to obtain the mixture 18 (13.5 g, 20% de ((2′S, 4′R) : (2′R, 4′S)=60:40)) as a colorless solid.
Mixture 19 of methyl 6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(n-butyl)piperidine-2′-carbonyl)-α-thiolincosaminide and methyl 6-N-((2′R, 4′S)-1′-N-(tert-butoxycarbonyl)-4′-(n-butyl)piperidine-2′-carbonyl)-α-thiolincosaminide
Compound (±)-6 (12.6 g, 44.2 mmol), 1-hydroxybenzotriazole (7.77 g, 57.5 mmol), N,N′-dicyclohexylcarbodiimide (11.0 g, 53.3 mmol) and MTL (14.6 g, 57.5 mmol) in DMF (120 ml) were treated for 20 h according to the similar procedure as described for the preparation of mixture 18 to afford 6-N-(1′-N-(tert-butoxycarbonyl)-4′-(n-butyl)piperidine-2′-carbonyl)-α-thiolincosaminide (20.0 g, 87%, (2′S, 4′R) isomer:(2′R, 4′S) isomer=ca. 50:50) as a colorless solid. To this colorless solid (14.53 g) was added ethyl acetate, and insoluble matter was filtrated off and ethyl acetate solution was concentrated under reduced pressure to obtain the mixture 19 (8.15 g, 80% de ((2′S, 4′R) : (2′R, 4′S)=90:10)) as a colorless solid.
Mixture 20 of methyl 6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(i-butyl)piperidine-2′-carbonyl)-α-thiolincosaminide and methyl 6-N-((2′R, 4′S)-1′-N-(tert-butoxycarbonyl)-4′-(i-butyl)piperidine-2′-carbonyl)-α-thiolincosaminide
To a solution of (±)-7 (6.00 g, 21.0 mmol) in DMF (57 ml) were added 1-hydroxybenzotriazole (2.84 g, 21.0 mmol), N,N′-dicyclohexylcarbodiimide (5.20 g, 25.2 mmol) and MTL (5.40 g, 21.3 mmol), and stirred at room temperature for 6 h. To the mixture were added H2O and ethyl acetate, and then the mixture was filtrated. The desired compound was extracted with ethyl acetate, washed with saturated aqueous KHCO3 and then the organic phase was dried over MgSO4, filtrated and concentrated under reduced pressure. To the resulting residue was added ethyl acetate, and insoluble matter was filtrated off and ethyl acetate solution was concentrated under reduced pressure. To the resulting residue was added toluene, and insoluble matter was filtrated off and toluene solution was concentrated under reduced pressure to obtain the mixture 20 (5.7 g, 90% de ((2′S, 4′R) : (2′R, 4′S)=95:5)) as a colorless solid.
Mixture 21 of methyl 6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(cyclopropylmethyl)piperidine-2′-carbonyl)-α-thiolincosaminide and methyl 6-N-((2′R, 4′S)-1′-N-(tert-butoxycarbonyl)-4′-(cyclopropylmethyl)piperidine-2′-carbonyl)-α-thiolincosaminide
Compound (±)-12 (44.2 g, 156 mmol), 1-hydroxybenzotriazole monohydrate (28.6 g, 187 mmol), N,N′-dicyclohexylcarbodiimide (35.8 g, 174 mmol) and MTL (47.4 g, 187 mmol) in DMF (300 ml) were treated for 13 h. To the mixture were added ethyl acetate and acetone, and then the solution was filtrated and concentrated under reduced pressure. To the resulting residue were added ethyl acetate, and the organic layer was washed with saturated aqueous NaHCO3 and saturated aqueous NaCl, and then filtrated and concentrated under reduced pressure. To the resulting residue was added ethyl acetate, and insoluble matter was filtrated off and ethyl acetate solution was concentrated under reduced pressure. To the resulting residue was added toluene, and insoluble matter was filtrated off and toluene solution was concentrated under reduced pressure to obtain the mixture 21 (36 g, 80% de ((2′S, 4′R) : (2′R, 4′S)=90:10)) as a colorless solid. FAB–MS m/z 519 (M+H)+ as C24H42N2O8S.
Methyl 6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(n-propyl)piperidine-2′-carbonyl)-2,3,4-tris-O-(trimethylsilyl)-α-thiolincosaminide (22)
To a solution of mixture 18 (13.5 g, 26.6 mmol, 20% de ((2′S, 4′R) : (2′R, 4′S)=60:40)) in pyridine (50 ml) were added trimethylchlorosilane (17.0 ml, 133 mmol) and hexamethyldisilazane (27.9 ml, 133 mmol), and stirred at room temperature for 40 min, then the solution was added to saturated aqueous NaHCO3. The desired compound was extracted with ethyl acetate, washed with saturated aqueous NaCl and then the organic phase was dried over Na2SO4, filtrated and concentrated under reduced pressure. To the resulting residue were added methanol (138 ml) and 6 N acetic acid (5.8 ml), and stirred at room temperature for 2.5 h. The mixture was added to saturated aqueous NaHCO3 and concentrated under reduced pressure to remove MeOH. The desired compound was extracted with ethyl acetate, and then the organic phase was dried over Na2SO4, filtrated and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane/ethyl acetate=19/1 to 3/1) to obtain the title compound (9.28 g, 48% (80% based on (2′S, 4′R) isomer) in 2 steps from mixture 18) as a colorless solid. ESI–MS m/z 723 (M+H)+ as C32H66N2O8SSi3; 1H NMR (400 MHz, CD3OD) δ 0.14 (s, 9 H), 0.16 (s, 9 H), 0.20 (s, 9 H), 0.90 (t, J=7.0 Hz, 3 H), 1.16 (d, J=6.2 Hz, 3 H), 1.22-1.39 (m, 4 H), 1.40–1.49 (m, 1 H), 1.46 (s, 9 H), 1.52–1.74 (m, 2 H), 1.75–1.87 (m, 1 H), 1.90–2.01 (m, 1 H), 2.04 (s, 3 H), 3.41–3.61 (m, 2 H), 3.75 (dd, J=9.6, 2.5 Hz, 1 H), 3.78–3.87 (m, 1 H), 4.07–4.20 (m, 3 H), 4.23–4.32 (m, 2 H), 5.17 (d, J=5.4 Hz, 1 H).
Methyl 6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(n-butyl)piperidine-2′-carbonyl)-2,3,4-tris-O-(trimethylsilyl)-α-thiolincosaminide (23)
Mixture 19 (8.15g, 15.7 mmol, 80% de ((2′S, 4′R) : (2′R, 4′S)=90:10), trimethylchlorosilane (100 ml, 78.3 mmol) and hexamethyldisilazane (16.4 ml, 78.3 mmol) in pyridine (30 ml) were treated for 20 min according to the similar procedure as described for the preparation of 22, and then the crude compound and 6 N acetic acid (3.4 ml) in MeOH (88 ml) were treated for 40 min according to the similar procedure as described for the preparation of 22 to afford 23 (8.20 g, 71% (79% based on (2′S, 4′R) isomer) in 2 steps from mixture 19) as a colorless solid. ESI–MS m/z 737 (M+H)+ as C33H68N2O8SSi3; 1H NMR (400 MHz, CDCl3) δ 0.14 (s, 18 H), 0.19 (s, 9 H), 0.82-0.93 (m, 3 H), 1.16 (d, J=6.6 Hz, 3 H), 1.18–1.36 (m, 7 H), 1.38–1.55 (m, 2 H), 1.46 (s, 9 H), 1.77–1.89 (m, 1 H), 2.00–2.10 (m, 1 H), 2.06 (s, 3 H), 2.90–3.03 (m, 1 H), 3.30–3.54 (m, 2 H), 3.60 (dd, J=9.6, 2.6 Hz, 1 H), 3.85–3.92 (m, 1 H), 3.94–4.07 (m, 2 H), 4.07–4.17 (m, 1 H), 4.26–4.40 (m, 1 H), 5.17 (d, J=5.4 Hz, 1 H), 6.32 (br d, J=9.0 Hz, 1 H).
Methyl 6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(i-butyl)piperidine-2′-carbonyl)-2,3,4-tris-O-(trimethylsilyl)-α-thiolincosaminide (24)
Mixture 20 (5.70 g, 10.9 mmol, 90% de ((2′S, 4′R) : (2′R, 4′S)=95:5), trimethylchlorosilane (13.2 ml, 103.6 mmol) and hexamethyldisilazane (21.7 ml, 104 mmol) in pyridine (21 ml) were treated for 1 h according to the similar procedure as described for the preparation of 22, and then the crude compound and 6 N acetic acid (2.4 ml) in MeOH (61 ml) were treated for 6 h according to the similar procedure as described for the preparation of 22 to afford 24 (7.25 g, 90% (95% based on (2′S, 4′R) isomer) in 2 steps from mixture 20) as a colorless solid. ESI–MS m/z 737 (M+H)+ as C33H68N2O8SSi3; 1H NMR (400 MHz, CD3OD) δ 0.14 (s, 9 H), 0.16 (s, 9 H), 0.20 (s, 9 H), 0.88 (d, J=6.5 Hz, 6 H), 1.08–1.33 (m, 3 H), 1.17 (d, J=6.4 Hz, 3 H), 1.46 (s, 9 H), 1.53–1.73 (m, 3 H), 1.77–1.88 (m, 1 H), 1.92–2.01 (m, 1 H), 2.05 (s, 3 H), 3.44–3.58 (m, 2 H), 3.74 (dd, J=9.6, 2.6 Hz, 1 H), 3.78–3.88 (m, 1 H), 4.05–4.17 (m, 3 H), 4.23–4.32 (m, 2 H), 5.17 (d, J=5.4 Hz, 1 H).
Methyl 6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(cyclopropylmethyl)piperidine-2′-carbonyl)-2,3,4-tris-O-(trimethylsilyl)-α-thiolincosaminide (25)
Mixture 21 (35.0 g, 67.5 mmol, 80% de ((2′S, 4′R) : (2′R, 4′S)=90:10), trimethylchlorosilane (43.1 ml, 337 mmol) and hexamethyldisilazane (70.6 ml, 337 mmol) in pyridine (130 ml) were treated for 1 h according to the similar procedure as described for the preparation of 22, and then the crude compound and 6 N acetic acid (16.9 ml) in MeOH (350 ml) were treated for 140 min according to the similar procedure as described for the preparation of 22 to afford 25 (30.6 g, 62% (69% based on (2′S, 4′R) isomer) in 2 steps from mixture 21) as a colorless solid. FAB–MS m/z 735 (M+H)+ as C33H66N2O8SSi3; 1H NMR (400 MHz, CD3OD) δ −0.04 to 0.04 (m, 2 H), 0.13 (s, 9 H), 0.16 (s, 9 H), 0.20 (s, 9 H), 0.40–0.49 (m, 2 H), 0.65–0.78 (m, 1 H), 1.17 (d, J=6.2 Hz, 3 H), 1.17–1.35 (m, 2 H), 1.34–1.47 (m, 1 H), 1.47 (s, 9 H), 1.64–1.91 (m, 3 H), 1.98–2.10 (m, 1 H), 2.05 (s, 3 H), 3.40–3.66 (m, 2 H), 3.75 (dd, J=9.7, 2.6 Hz, 1 H), 3.76–3.85 (m, 1 H), 4.12 (dd, J=9.7, 5.4 Hz, 1 H), 4.13–4.19 (m, 1 H), 4.20–4.35 (m, 3 H), 5.18 (d, J=5.4 Hz, 1 H).
Methyl (7S)-7-acetylthio-6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(n-propyl)piperidine-2′-carbonyl)-7-deoxy-2,3,4-tris-O-(trimethylsilyl)-α-thiolincosaminide (26)
To a solution of 22 (500 mg, 0.69 mmol) in CH2Cl2 (2 ml) at 0 °C were added Et3N (291 μl, 2.08 mmol) and methanesulfonyl chloride (107 μl, 1.38 mmol), and stirred at room temperature for 1 h. The mixture was added to saturated aqueous NaHCO3, extracted with ethyl acetate, dried over Na2SO4 and concentrated under reduced pressure to obtain methyl 6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(n-propyl)piperidine-2′-carbonyl)-7-O-methanesulfonyl-2,3,4-tris-O-(trimethylsilyl)-α-thiolincosaminide as a crude compound (530 mg). To a solution of this crude compound (530 mg) in DMF (3.0 ml) was added AcSK (396 mg, 3.47 mmol) and stirred at 80 °C for 2 h. The mixture was added to saturated aqueous NaHCO3, then extracted with ethyl acetate, washed with 10% aqueous NaCl, dried over Na2SO4, filtrated and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane to hexane/ethyl acetate=85/15) to obtain the title compound (357 mg, 66% in 2 steps from 22) as a colorless solid. FAB–MS m/z 781 (M+H)+ as C34H68N2O8S2Si3; 1H NMR (400 MHz, CDCl3) δ 0.126 (s, 9 H), 0.13 (s, 9 H), 0.18 (s, 9 H), 0.88 (t, J=6.9 Hz, 3 H), 1.10–1.21 (m, 1 H), 1.23–1.38 (m, 4 H), 1.35 (d, J=6.8 Hz, 3 H), 1.42–1.60 (m, 2 H), 1.49 (s, 9 H), 1.80–1.94 (m, 1 H), 1.95–2.08 (m, 1 H), 1.99 (s, 3 H), 2.29 (s, 3 H), 3.00–3.18 (m, 1 H), 3.57 (dd, J=9.5, 2.2 Hz, 1 H), 3.63-3.83 (m, 2 H), 3.87–4.04 (m, 2 H), 4.13 (dd, J=9.5, 5.6 Hz, 1 H), 4.22–4.33 (m, 1 H), 4.50–4.62 (m, 1 H), 5.15 (d, J=5.6 Hz, 1 H), 6.04–6.37 (m, 1 H).
Methyl (7S)-7-acetylthio-6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(n-butyl)piperidine-2′-carbonyl)-7-deoxy-2,3,4-tris-O-(trimethylsilyl)-α-thiolincosaminide (27)
Compound 23 (1.01 g, 1.37 mmol), Et3N (490 μl, 3.48 mmol) and methanesulfonyl chloride (220 μl, 2.79 mmol) in CH2Cl2 (20 ml) at 0 °C were treated for 1 h according to the similar procedure as described for the preparation of 26, and then a crude mesylate (1.17 g) and AcSK (992 mg, 8.68 mmol) in DMF (13 ml) at 80 °C were treated for 3 h according to the similar procedure as described for the preparation of 26 to afford 27 (565 mg, 52% in 2 steps from 23) as a colorless solid. FAB–MS m/z 795 (M+H)+ as C35H70N2O8S2Si3; 1H NMR (400 MHz, CDCl3) δ 0.11 (s, 9 H), 0.12 (s, 9 H), 0.17 (s, 9 H), 0.78–0.95 (m, 3 H), 1.07–1.32 (m, 7 H), 1.34 (d, J=6.9 Hz, 3 H), 1.38–1.60 (m, 2 H), 1.48 (s, 9 H), 1.78–1.95 (m, 1 H), 1.95–2.11 (m, 1 H), 1.97 (s, 3 H), 2.27 (s, 3 H), 3.00–3.18 (m, 1 H), 3.54 (dd, J=9.6, 2.2 Hz, 1 H), 3.62–3.85 (m, 2 H), 3.85–4.05 (m, 2 H), 4.06–4.16 (m, 1 H), 4.21-4.31 (m, 1 H), 4.50–4.62 (m, 1 H), 5.14 (d, J=5.5 Hz, 1 H), 6.05–6.48 (m, 1 H).
Methyl (7S)-7-acetylthio-6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(i-butyl)piperidine-2′-carbonyl)-7-deoxy-2,3,4-tris-O-(trimethylsilyl)-α-thiolincosaminide (28)
Compound 24 (1.01 g, 1.37 mmol), Et3N (930 μl, 6.85 mmol) and methanesulfonyl chloride (210 μl, 2.74 mmol) in CH2Cl2 (20 ml) at 0 °C were treated for 1 h according to the similar procedure as described for the preparation of 26, and then the crude mesylate (1.17 g) and AcSK (470 mg, 4.11 mmol) in DMF (6 ml) at 80 °C were treated for 3 h according to the similar procedure as described for the preparation of 26 to afford 28 (598 mg, 55% in 2 steps from 24) as a colorless solid.
Methyl (7S)-7-acetylthio-6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(cyclopropylmethyl)piperidine-2′-carbonyl)-7-deoxy-2,3,4-tris-O-(trimethylsilyl)-α-thiolincosaminide (29)
Compound 25 (7.40 g, 10.1 mmol), Et3N (4.24 ml, 30.3 mmol) and methanesulfonyl chloride (1.56 ml, 20.2 mmol) in CHCl3 (70 ml) at 0 °C were treated and then the solution was stirred at room temperature for 1 h according to the similar procedure as described for the preparation of 26 and then, the crude mesylate and AcSK (5.76 g, 50.5 mmol) in DMF (75 ml) at 80 °C were treated for 1.5 h according to the similar procedure as described for the preparation of 26 to afford 29 (4.30 g, 54% in 2 steps from 25) as a colorless solid. ESI–MS m/z 793 (M+H)+ as C35H68N2O8S2Si3; 1H NMR (400 MHz, CDCl3) δ −0.05 to 0.03 (m, 2 H), 0.125 (s, 9 H), 0.131 (s, 9 H), 0.18 (s, 9 H), 0.38–0.45 (m, 2 H), 0.60-0.73 (m, 1 H), 1.17–1.29 (m, 3 H), 1.35 (d, J=6.8 Hz, 3 H), 1.50 (s, 9 H), 1.59–1.72 (m, 2 H), 1.86–1.97 (m, 1 H), 2.00 (s, 3 H), 2.06–2.14 (m, 1 H), 2.29 (s, 3 H), 3.03–3.18 (m, 1 H), 3.58 (dd, J=9.6, 2.3 Hz, 1 H), 3.67–3.84 (m, 2 H), 3.88–4.02 (m, 2 H), 4.12 (dd, J=9.5, 5.4 Hz, 1 H), 4.30–4.38 (m, 1 H), 4.52–4.60 (m, 1 H), 5.16 (d, J=5.4 Hz, 1 H), 6.10–6.45 (m, 1 H).
Methyl (7S)-6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(n-propyl)piperidine-2′-carbonyl)-7-deoxy-7-mercapto-α-thiolincosaminide (30)
To a solution of 26 (341 mg, 0.436 mmol) in MeOH (4 ml) was added 1 N HCl (2.5 ml) and stirred at room temperature for 5 min. The mixture was added to saturated aqueous NaHCO3, concentrated under reduced pressure to remove MeOH until half volume, extracted with ethyl acetate, dried over Na2SO4 and concentrated under reduced pressure to obtain methyl (7S)-7-acetylthio-6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(n-propyl)piperidine-2′-carbonyl)-7-deoxy-α-thiolincosaminide (246.5 mg, quant) as a colorless solid. To a solution of this intermediate (244 mg, 0.432 mmol) in MeOH (2.5 ml) was added 28% NaOMe/MeOH solution (251 μl, 1.30 mmol), stirred at room temperature for 20 min. The mixture was added to a saturated aqueous NH4Cl, extracted with ethyl acetate, washed with saturated aqueous NaHCO3, dried over Na2SO4 and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane to hexane/ethyl acetate=96/4) to obtain the title compound (234 mg, 96%) as a colorless solid. ESI–MS m/z 523 (M+H)+ as C23H42N2O7S2; 1H NMR (400 MHz, CD3OD) δ 0.92 (t, J=7.0 Hz, 3 H), 1.24–1.41 (m, 5 H), 1.30 (d, J=7.0 Hz, 3 H), 1.46 (s, 9 H), 1.52–1.64 (m, 2 H), 1.77–1.90 (m, 1 H), 1.94–2.06 (m, 1 H), 2.15 (s, 3 H), 3.40–3.64 (m, 2 H), 3.46 (dq, J=7.0, 2.4 Hz, 1 H), 3.54 (dd, J=10.3, 3.4 Hz, 1 H), 3.94–4.01 (m, 1 H), 4.06 (dd, J=10.3, 5.6 Hz, 1 H), 4.18–4.26 (m, 2 H), 4.32 (dd, J=9.8, 2.4 Hz, 1 H), 5.25 (d, J=5.6 Hz, 1 H).
Methyl (7S)-6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(n-butyl)piperidine-2′-carbonyl)-7-deoxy-7-mercapto-α-thiolincosaminide (31)
To a solution of 27 (565 mg, 0.711 mmol) in MeOH (5.6 ml) was added 1 N HCl (5.6 ml) and stirred at room temperature for 100 min. The mixture was added to 8% aqueous NaHCO3, extracted with ethyl acetate, washed with 25% aqueous NaCl, dried over Na2SO4 and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (CHCl3/MeOH=30/1) to obtain methyl (7S)-7-acetylthio-6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(n-butyl)piperidine-2′-carbonyl)-7-deoxy-α-thiolincosaminide (378 mg, 91.8%) as a colorless solid. To a solution of this intermediate (378 mg, 0.652 mmol) in MeOH (4 ml) was added NaOMe (115 mg, 2.02 mmol) and stirred at room temperature for 3 h. The mixture was added to 8% aqueous NaHCO3, extracted with ethyl acetate, washed with 25% aqueous NaCl, dried over Na2SO4 and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (CHCl3/MeOH=40/1) to obtain the title compound (373 mg, quant) as a colorless solid. ESI–MS m/z 537 (M+H)+ as C24H44N2O7S2; 1H NMR (400 MHz, CD3OD) δ 0.87–0.95 (m, 3 H), 1.23-1.40 (m, 7 H), 1.30 (br d, J=7.2 Hz, 3 H), 1.46 (s, 9 H), 1.50–1.65 (m, 2 H), 1.77–1.89 (m, 1 H), 1.96–2.06 (m, 1 H), 2.15 (s, 3 H), 3.42–3.60 (m, 2 H), 3.45 (dq, J=7.1, 2.3 Hz, 1 H), 3.54 (dd, J=10.3, 3.4 Hz, 1 H), 3.94–4.00 (m, 1 H), 4.06 (dd, J=10.1, 5.6 Hz, 1 H), 4.10–4.26 (m, 2 H), 4.32 (dd, J=9.8, 2.3 Hz, 1 H), 5.24 (d, J=5.6 Hz, 1 H).
Methyl (7S)-6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(i-butyl)piperidine-2′-carbonyl)-7-deoxy-7-mercapto-α-thiolincosaminide (32)
To a solution of 28 (565 mg, 0.711 mmol) in MeOH (5.6 ml) was added 5 N HCl (0.3 ml) and stirred at room temperature for 30 min. The mixture was added to 8% aqueous NaHCO3, extracted with ethyl acetate, washed with 25% aqueous NaCl, dried over Na2SO4 and concentrated under reduced pressure to obtain methyl (7S)-7-acetylthio-6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(i-butyl)piperidine-2′-carbonyl)-7-deoxy-α-thiolincosaminide (410 mg) as a crude compound. Then, the crude compound (410 mg) in MeOH (13 ml) was added and 5 N NaOMe (430 μl, 2.15 mmol) in MeOH at room temperature were treated for 1 h according to the similar procedure as described for the preparation of 31 to afford 32 (362 mg, 88% in 2 steps from 28) as a colorless solid. ESI–MS m/z 537 (M+H)+ as C24H44N2O7S2; 1H NMR (400 MHz, CD3OD) δ 0.89 (d, J=2.8 Hz, 3 H), 0.90 (d, J=2.9 Hz, 3 H), 1.12–1.27 (m, 3 H), 1.30 (d, J=7.1 Hz, 3 H), 1.46 (s, 9 H), 1.49–1.60 (m, 1 H), 1.60–1.74 (m, 2 H), 1.75–1.88 (m, 1 H), 1.95–2.04 (m, 1 H), 2.15 (s, 3 H), 3.40–3.59 (m, 2 H), 3.45 (dq, J=7.1, 2.3 Hz, 1 H), 3.54 (dd, J=10.2, 3.4 Hz, 1 H), 3.93–4.02 (m, 1 H), 4.06 (dd, J=10.2, 5.6 Hz, 1 H), 4.11–4.26 (m, 2 H), 4.32 (dd, J=9.8, 2.3 Hz, 1 H), 5.25 (d, J=5.6 Hz, 1 H).
Methyl (7S)-6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(cyclopropylmethyl)piperidine-2′-carbonyl)-7-deoxy-7-mercapto-α-thiolincosaminide (33)
To a solution of 29 (5.20 g, 6.55 mmol) in MeOH (70 ml) was added 1 N HCl (26.2 ml) and stirred at room temperature for 5 min. The mixture was added to 10% aqueous NaHCO3 and then concentrated under reduced pressure until half volume to remove MeOH, extracted with ethyl acetate, washed with 25% aqueous NaCl, dried over Na2SO4 and concentrated under reduced pressure to obtain methyl (7S)-7-acetylthio-6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(cyclopropylmethyl)piperidine-2′-carbonyl)-7-deoxy-α-thiolincosaminide (4.70 g) as a crude compound. Then, the crude compound (4.70 g) and 28% NaOMe/MeOH solution (3.79 ml, 1.06 mmol) in MeOH (38 ml) at room temperature were treated for 15 min according to the similar procedure as described for the preparation of 30 to afford 33 (3.45 g, 99% in 2 steps from 29) as a colorless solid. FAB–MS m/z 535 (M+H)+ as C24H42N2O7S2; 1H NMR (400 MHz, CD3OD) δ 0.01–0.06 (m, 2 H), 0.39–0.49 (m, 2 H), 0.63–0.78 (m, 1 H), 1.16–1.28 (m, 2 H), 1.30 (d, J=7.1 Hz, 3 H), 1.34–1.43 (m, 1 H), 1.47 (s, 9 H), 1.55–1.76 (m, 2 H), 1.82–1.93 (m, 1 H), 2.05–2.14 (m, 1 H), 2.15 (s, 3 H), 3.40–3.66 (m, 2 H), 3.45 (dq, J=7.1, 2.4 Hz, 1 H), 3.54 (dd, J=10.3, 3.4 Hz, 1 H), 3.94–4.01 (m, 1 H), 4.06 (dd, J=10.3, 5.6 Hz, 1 H), 4.13–4.25 (m, 2 H), 4.32 (dd, J=9.7, 2.4 Hz, 1 H), 5.25 (d, J=5.6 Hz, 1 H).
Methyl 6-N-((2′S, Z)-1′-N-(2′′-nitrophenylsulfonyl)-5′-n-propyl-2′,3′,6′,7′-tetrahydro-1H-azepine-2′-carbonyl)-α-thiolincosaminide (34)
To a solution of 17 (481.5 mg, 1.31 mmol) in DMF (5 ml) were added 1-hydroxybenzotriazole (265.0 mg, 1.96 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (375.8 mg, 1.96 mmol) and MTL (496.8 mg, 1.96 mmol), and stirred at room temperature for 14 h. To the mixture were added ethyl acetate and saturated aqueous NaHCO3. The desired compound was extracted with ethyl acetate, washed with H2O and then the organic phase was dried over Na2SO4, filtrated and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (CHCl3/MeOH=50/1 to 30/1) to obtain the title compound (660 mg, 84%) as a colorless solid. FAB–MS m/z 604 (M+H)+ as C25H37N3O10S2; 1H NMR (400 MHz, CD3OD) δ 0.83 (t, J=7.3 Hz, 3 H), 1.15 (d, J=6.3 Hz, 3 H), 1.25–1.37 (m, 2 H), 1.08 (br t, J=7.4 Hz, 2 H), 2.05 (s, 3 H), 2.23–2.56 (m, 3 H), 2.68–2.80 (m, 1 H), 3.58 (dd, J=10.2, 3.4 Hz, 1 H), 3.73–3.88 (m, 3 H), 4.02–4.12 (m, 3 H), 4.36–4.41 (m, 1 H), 4.74 (dd, J=8.0, 3.7 Hz, 1 H), 5.22 (d, J=5.6 Hz, 1 H), 5.39 (br t, J=6.3 Hz, 1 H), 7.74–7.86 (m, 3 H), 8.09–8.17 (m, 1 H).
Methyl 6-N-((2′S)-5′-n-propylazepane-2′-carbonyl)-α-thiolincosaminide (35) (stereochemistry at the C-5′ position is not assigned)
To a solution of 34 (1.15 g, 1.90 mmol) in DMF (10 ml) at 0 °C were added 4-bromobenzenethiol (721 mg, 3.81 mmol) and cesium carbonate (1.25 g, 3.84 mmol), and then stirred at room temperature for 2 h. The mixture was concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane to hexane/ethyl acetate=50/50, then CHCl3/CH3OH/28% aq NH4OH=20/1/0.1) to obtain methyl 6-N-((2′S, Z)-5′-n-propyl-2′,3′,6′,7′-tetrahydro-1H-azepine-2′-carbonyl)-α-thiolincosaminide (649.1 mg, 81.4%) as a colorless solid. To this intermediate (649.1 mg, 1.55 mmol) in MeOH (30 ml) was added Pd/C (324 mg) and then vigorously stirred in hydrogen atmosphere of 0.95 MPa at 40 °C for 3.5 h. The mixture was filtrated off with celite and the mother liquor was concentrated under reduced pressure. To the resulting residue were added Pd/C (324 mg) and MeOH (30 ml), and then the mixture was vigorously stirred in hydrogen atmosphere of 0.95 MPa at 40 °C for 65 h. The mixture was filtrated with celite and concentrated under reduced pressure to obtain the title compound (560 mg, 86%) as a colorless solid. FAB–MS m/z 421 (M+H)+ as C19H36N2O6S; TOF–ESI–HR-MS (M+H)+ calcd for C19H36N2O6S: 421.2372, found: 421.2370; 1H NMR (400 MHz, CD3OD) δ 0.90 (t, J=7.2 Hz, 3 H), 1.18 (d, J=6.6 Hz, 3 H), 1.20–1.48 (m, 7 H), 1.59–1.71 (m, 1 H), 1.77–1.88 (m, 1 H), 1.93–2.04 (m, 2 H), 2.08 (s, 3 H), 2.73–2.84 (m, 1 H), 3.03 (ddd, J=13.8, 5.5, 2.1 Hz, 1 H), 3.53–3.62 (m, 2 H), 3.94–4.05 (m, 2 H), 4.10 (dd, J=10.2, 5.6 Hz, 1 H), 4.15–4.20 (m, 1 H), 4.22–4.26 (m, 1 H), 5.24 (d, J=5.4 Hz, 1 H).
Methyl 6-N-((2′ S )-1′-N-(tert-butoxycarbonyl)-5′- n -propylazepane-2-carbonyl)-α-thiolincosaminide (36) (stereochemistry at the C-5′ position is not assigned)
To a solution of 35 (560 mg, 1.34 mmol) in 1,4-dioxane (10 ml)-H2O (10 ml) were added LiOH·H2O (84.0 mg, 2.00 mmol) and di-tert-butyl dicarbonate (0.37 ml, 1.6 mmol), and then stirred at room temperature for 3 h. The mixture was added to saturated aqueous NaHCO3, extracted with ethyl acetate and then the organic phase was dried over Na2SO4, filtrated and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (CHCl3/CH3OH/28% aq NH4OH=20/1/0.1) to obtain the title compound (483.3 mg, 70%) as a colorless solid. ESI–MS m/z 521 (M+H)+ as C24H44N2O8S; 1H NMR (400 MHz, CD3OD) δ 0.85–0.96 (m, 3 H), 1.12–1.38 (m, 7 H), 1.47 (s, 9 H), 1.50–1.65 (m, 4 H), 1.66–1.80 (m, 1 H), 1.84–2.14 (m, 2 H), 2.07 (s, 3 H), 3.43–3.55 (m, 2 H), 3.59 (dd, J=10.1, 3.3 Hz, 1 H), 3.75–3.90 (m, 1 H), 3.98–4.18 (m, 3 H), 4.26–4.45 (m, 2 H), 5.24 (d, J=5.6 Hz, 1 H).
Methyl 6-N-((2′ S )-1′-N-(tert-butoxycarbonyl)-5′- n -propylazepane-2-carbonyl)-2,3,4-tris-O-(trimethylsilyl)-α-thiolincosaminide (37) (stereochemistry at the C-5′ position is not assigned)
Compound 36 (894.0 mg, 1.72 mmol), trimethylchlorosilane (1.09 ml, 8.59 mmol) and hexamethyldisilazane (1.80 ml, 8.59 mmol) in pyridine (10 ml) were treated for 30 min according to the similar procedure as described for the preparation of 22, and then the crude fully protected intermediate and 2 N acetic acid (2.23 ml) in MeOH (36 ml) were stirred at room temperature for 1 h. The mixture was added to saturated aqueous NaHCO3, extracted with ethyl acetate and then the organic phase was dried over Na2SO4, filtrated and concentrated under reduced pressure to afford 37 as a crude compound.
Methyl (7S)-7-acetylthio-6-N-((2′ S )-1′-N-(tert-butoxycarbonyl)-5′- n -propylazepane-2-carbonyl)-7-deoxy-2,3,4-tris-O-(trimethylsilyl)-α-thiolincosaminide (38) (stereochemistry at the C-5′ position is not assigned)
Crude 37, Et3N (1.20 ml, 8.58 mmol) and methanesulfonyl chloride (0.53 ml, 6.9 mmol) in CHCl3 (20 ml) were treated at room temperature for 1 h according to the similar procedure as described for the preparation of 26, and then the crude mesylate and AcSK (1.19 g, 10.4 mmol) in DMF (9.8 ml) at 80 °C were treated for 2 h according to the similar procedure as described for the preparation of 26 to afford 38 (768.6 mg, 56% in 4 steps from 36) as a colorless solid. FAB–MS m/z 795 (M+H)+ as C35H70N2O8S2Si3.
Methyl 6-N-((2′ S )-1′-N-(tert-butoxycarbonyl)-5′- n -propylazepane-2-carbonyl)-(7S)-7-mercapto-α-thiolincosaminide (39) (stereochemistry at the C-5′ position is not assigned)
To a solution of 38 (768.6 mg, 0.966 mmol) in MeOH (16 ml) at 0 °C was added 1 N HCl (1.6 ml) and then the mixture was stirred at 0 °C for 30 min. The mixture was concentrated under reduced pressure to obtain methyl (7S)-7-acetylthio-6-N-((2′S)-1′-N-(tert-butoxycarbonyl)-5′-n-propylazepane-2-carbonyl)-7-deoxy-α-thiolincosaminide as a crude compound and then, the crude intermediate and 28% NaOMe/MeOH soultion (0.238 ml, 0.966 mmol) in MeOH (16 ml) at room temperature were treated for 30 min according to the similar procedure as described for the preparation of 30 to afford 39 (170 mg, 33% in 2 steps from 38) as a colorless solid. ESI–MS m/z 537 (M+H)+ as C24H44N2O7S2; TOF–ESI–HR-MS (M+H)+ calcd for C24H44N2O7S2: 537.2668, found: 537.2668; 1H NMR (400 MHz, CD3OD) δ 0.92 (br t, J=6.6 Hz, 3 H), 1.20–1.80 (m, 12 H), 1.48 (s, 9 H), 1.88–2.07 (m, 2 H), 2.15 (s, 3 H), 3.38–3.74 (m, 4 H), 3.76–3.94 (m, 1 H), 4.00–4.12 (m, 2 H), 4.23–4.34 (m, 1 H), 4.36–4.56 (m, 1 H), 5.24 (d, J=5.6 Hz, 1 H).
Methyl (7S)-6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(n-propyl)piperidine-2′-carbonyl)-7-deoxy-7-(4-(pyrimidin-5-yl)phenylthio)-α-thiolincosaminide (40)
To a solution of 5-(4-bromophenyl)pyrimidine (103.4 mg, 0.440 mmol), 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (Xantphos) (18.8 mg, 0.0315 mmol) and tris(dibenzylideneacetone)dipalladium(0) (Pd2(dba)3) (13.4 mg, 0.0146 mmol) in 1,4-dioxane (2.0 ml) were added compound 30 (152.1 mg, 0.291 mmol) and N,N-diisopropylethylamine (0.100 ml, 0.576 mmol), and refluxed for 6 h. The mixture was filtrated by either Chromatodisc (0.45 μm) (Kurabo Industries Ltd, Osaka, Japan) or celite, concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (CHCl3/MeOH/28% aq NH4OH=10/1/0.1) to obtain the title compound (163.0 mg, 83%) as an off white solid. FAB–MS m/z 677 (M+H)+ as C33H48N4O7S2.
Methyl (7S)-6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(n-butyl)piperidine-2′-carbonyl)-7-deoxy-7-(4-(pyrimidin-5-yl)phenylthio)-α-thiolincosaminide (41)
Compound 31 (150.1 mg, 0.280 mmol), 5-(4-bromophenyl)pyrimidine (99.3 mg, 0.422 mmol), Xantphos (17.3 mg, 0.029 mmol), Pd2(dba)3 (13.4 mg, 0.0146 mmol) and N,N-diisopropylethylamine (97.0 μl, 0.559 mmol) in 1,4-dioxane (2.0 ml) were treated for 6 h according to the similar procedure as described for the preparation of 40 to afford 41 (150.8 mg, 78%) as a colorless solid.
Methyl (7S)-6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(i-butyl)piperidine-2′-carbonyl)-7-deoxy-7-(4-(pyrimidin-5-yl)phenylthio)-α-thiolincosaminide (42)
Compound 32 (100 mg, 0.186 mmol), 5-(4-bromophenyl)pyrimidine (50.0 mg, 0.213 mmol), Xantphos (10.0 mg, 0.0173 mmol), Pd2(dba)3 (10.0 mg, 0.0109 mmol) and N,N-diisopropylethylamine (60.0 μl, 0.353 mmol) in 1,4-dioxane (1.3 ml) were treated for 6 h according to the similar procedure as described for the preparation of 40 to afford 42 (105.0 mg, 82%) as a colorless solid.
Methyl (7S)-6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(cyclopropylmethyl)piperidine-2′-carbonyl)-7-deoxy-7-(4-(pyrimidin-5-yl)phenylthio)-α-thiolincosaminide (43)
Compound 33 (1.35 g, 2.52 mmol), 5-(4-bromophenyl)pyrimidine (771 mg, 3.28 mmol), Xantphos (151 mg, 0.253 mmol), Pd2(dba)3 (117 mg, 0.128 mmol) and N,N-diisopropylethylamine (0.875 ml, 5.04 mmol) in 1,4-dioxane (20 ml) were treated for 6 h according to the similar procedure as described for the preparation of 40 to afford 43 (1.63 g, 93%) as an off white solid. ESI–MS m/z 689 (M+H)+ as C34H48N4O7S2.
Methyl (7S)-6-N-((2′S, 4′R)-4′-(n-propyl)piperidine-2′-carbonyl)-7-deoxy-7-(4-(pyrimidin-5-yl)phenylthio)-α-thiolincosaminide (44)
To a solution of 40 (163 mg, 0.241 mmol) in CH2Cl2 (3.3 ml) at −20 °C was added 2,2,2-trifluoroacetic acid (0.36 ml) and then the solution was stirred at room temperature for 4 h. The solution was concentrated under reduced pressure. The resulting residue was purified by preparative TLC (CHCl3/MeOH/28% aq NH4OH=9/2/0.2) to obtain the title compound (122.8 mg, 88%) as a colorless solid. [α]D22 +85.2° (c 1.02, MeOH); ESI–MS m/z 577 (M+H)+ as C28H40N4O5S2; TOF–ESI–HR-MS (M+H)+ calcd for C28H40N4O5S2: 577.2518, found: 577.2516; 1H NMR (400 MHz, CD3OD) δ 0.90 (t, J=7.3 Hz, 3 H), 1.02–1.15 (m, 2 H), 1.20–1.29 (m, 2 H), 1.31–1.42 (m, 2 H), 1.35 (d, J=6.8 Hz, 3 H), 1.49–1.63 (m, 1 H), 1.68–1.77 (m, 1 H), 1.93 (s, 3 H), 1.96–2.05 (m, 1 H), 3.19 (dt, J=10.9, 2.0 Hz, 1 H), 3.15-3.23 (m, 1 H), 3.40 (dd, J=11.9, 2.8 Hz, 1 H), 3.58 (dd, J=10.3, 3.3 Hz, 1 H), 3.88 (br dd, J=3.3, 0.8 Hz, 1 H), 3.93 (dq, J=6.8, 2.4 Hz, 1 H), 4.09 (dd, J=10.3, 5.5 Hz, 1 H), 4.44 (br dd, J=10.0, 0.8 Hz, 1 H), 4.61 (dd, J=10.0, 2.4 Hz, 1 H), 5.27 (d, J=5.5 Hz, 1 H), 7.52–7.58 (m, 2 H), 7.66–7.71 (m, 2 H), 9.06 (s, 2 H), 9.11 (s, 1 H).
Methyl (7S)-6-N-((2′S, 4′R)-4′-(n-butyl)piperidine-2′-carbonyl)-7-deoxy-7-(4-(pyrimidin-5-yl)phenylthio)-α-thiolincosaminide (45)
Compound 41 (150.8 mg, 0.218 mmol) and 2,2,2-trifluoroacetic acid (0.33 ml) in CH2Cl2 (3.0 ml) were treated at −20 °C for 10 min and then treated room temperature for 2.5 h according to the similar procedure as described for the preparation of 44 to afford 45 (102.6 mg, 80%) as a colorless solid. [α]D23 +81.2° (c 0.78, MeOH); ESI–MS m/z 591 (M+H)+ as C29H42N4O5S2; TOF–ESI–HR-MS (M+H)+ calcd for C29H42N4O5S2: 591.2675, found: 591.2669; 1H NMR (400 MHz, CD3OD) δ 0.85–0.96 (m, 3 H), 1.09–1.22 (m, 2 H), 1.23–1.41 (m, 6 H), 1.36 (d, J=6.8 Hz, 3 H), 1.51–1.66 (m, 1 H), 1.74–1.84 (m, 1 H), 1.93 (s, 3 H), 2.04-2.12 (m, 1 H), 2.78 (dt, J=12.9, 2.8 Hz, 1 H), 3.21–3.29 (m, 1 H), 3.52 (dd, J=12.1, 2.9 Hz, 1 H), 3.58 (dd, J=10.2, 3.3 Hz, 1 H), 3.89 (br dd, J=3.3, 0.8 Hz, 1 H), 3.93 (dq, J=6.8, 2.5 Hz, 1 H), 4.09 (dd, J=10.2, 5.6 Hz, 1 H), 4.45 (br dd, J=10.0, 0.8 Hz, 1 H), 4.63 (dd, J=10.0, 2.5 Hz, 1 H), 5.27 (d, J=5.6 Hz, 1 H), 7.51–7.58 (m, 2 H), 7.65–7.72 (m, 2 H), 9.06 (s, 2 H), 9.11 (s, 1 H).
Methyl (7S)-6-N-((2′S, 4′R)-4′-(i-butyl)piperidine-2′-carbonyl)-7-deoxy-7-(4-(pyrimidin-5-yl)phenylthio)-α-thiolincosaminide (46)
Compound 42 (100 mg, 0.145 mmol) and 2,2,2-trifluoroacetic acid (0.50 ml) were treated at 0 °C for 1 h according to the similar procedure as described for the preparation of 44 to afford 46 (55.0 mg, 64%) as a colorless solid. [α]D24 +93.9° (c 0.83, MeOH); ESI–MS m/z 591 (M+H)+ as C29H42N4O5S2; TOF–ESI–HR-MS (M+H)+ calcd for C29H42N4O5S2: 591.2675, found: 591.2674; 1H NMR (400 MHz, CD3OD) δ 0.91 (d, J=5.7 Hz, 3 H), 0.90 (d, J=5.7 Hz, 3 H), 1.08–1.20 (m, 3 H), 1.36 (d, J=7.0 Hz, 3 H), 1.64–1.82 (m, 4 H), 1.93 (s, 3 H), 2.03–2.12 (m, 1 H), 2.76-2.86 (m, 1 H), 3.21–3.28 (m, 1 H), 3.51–3.61 (m, 2 H), 3.89 (br dd, J=3.2, 0.8 Hz, 1 H), 3.93 (dq, J=7.0, 2.4 Hz, 1 H), 4.09 (dd, J=10.2, 5.6 Hz, 1 H), 4.45 (br dd, J=10.0, 0.8 Hz, 1 H), 4.63 (dd, J=10.0, 2.4 Hz, 1 H), 5.27 (d, J=5.6 Hz, 1 H), 7.52–7.57 (m, 2 H), 7.66–7.71 (m, 2 H), 9.06 (s, 2 H), 9.11 (s, 1 H).
Methyl (7S)-6-N-((2′S, 4′R)-4′-(cyclopropylmethyl)piperidine-2′-carbonyl)-7-deoxy-7-(4-(pyrimidin-5-yl)phenylthio)-α-thiolincosaminide (47)
Compound 43 (1.63 g, 2.36 mmol) and 2,2,2-trifluoroacetic acid (3.5 ml) in CH2Cl2 (32 ml) were treated at −20 °C for 20 min, and then treated room temperature for 5.5 h according to the similar procedure as described for the preparation of 44 to afford 47 (1.12 g, 81%) as a colorless solid. [α]D24 +86.1° (c 0.25, MeOH); ESI–MS m/z 589 (M+H)+ as C29H40N4O5S2; TOF–ESI–HR-MS (M+H)+ calcd for C29H40N4O5S2: 589.2518, found: 589.2517; IR (KBr) cm−1 1046, 1078, 1415, 1508, 1602, 1653, 1671, 1698, 2338, 2360, 3001, 3347 and 3690 cm−1; 1H NMR (400 MHz, CD3OD) δ −0.01 to 0.08 (m, 2 H), 0.43–0.52 (m, 2 H), 0.67–0.78 (m, 1 H), 1.14–1.23 (m, 1 H), 1.24–1.40 (m, 3 H), 1.36 (d, J=6.8 Hz, 3 H), 1.75-1.89 (m, 1 H), 1.90–2.00 (m, 1 H), 1.93 (s, 3 H), 2.28–2.37 (m, 1 H), 2.96 (dt, J=13.1, 3.0 Hz, 1 H), 3.33–3.40 (m, 1 H), 3.59 (dd, J=10.2, 3.1 Hz, 1 H), 3.75 (dd, J=12.4, 3.0 Hz, 1 H), 3.90 (br dd, J=3.1, 0.8 Hz, 1 H), 3.93 (dq, J=6.8, 2.5 Hz, 1 H), 4.10 (dd, J=10.2, 5.6 Hz, 1 H), 4.46 (br dd, J=10.0, 0.8 Hz, 1 H), 4.65 (dd, J=10.0, 2.5 Hz, 1 H), 5.28 (d, J=5.6 Hz, 1 H), 7.51–7.57 (m, 2 H), 7.65–7.73 (m, 2 H), 9.06 (s, 2 H), 9.11 (s, 1 H); 13C NMR (100 MHz, CD3OD) δ 5.1, 9.2, 13.8, 20.7, 33.8, 38.2, 38.8, 43.5, 44.8, 46.4, 53.8, 61.1, 69.6, 69.9, 71.0, 72.1, 90.2,128.6, 131.7, 133.0, 135.3, 138.6, 155.9, 157.9 and 176.5.
Methyl (7S)-6-N-((2′ S )-1′-N-(tert-butoxycarbonyl)-5′- n -propylazepane-2-carbonyl)-7-deoxy-7-(4-(pyrimidin-5-yl)phenylthio)-α-thiolincosaminide (48) (stereochemistry at the C-5′ position is not assigned)
Compound 39 (15.0 mg, 0.0279 mmol), 5-(4-bromophenyl)pyrimidine (13.1 mg, 0.0558 mmol), Xantphos (3.2 mg, 5.58 μmol), Pd2(dba)3 (5.1 mg, 5.6 μmol) and N,N-diisopropylethylamine (10.0 μl, 0.0558 mmol) in 1,4-dioxane (0.2 ml) were treated for 2 h according to the similar procedure as described for the preparation of 40 to afford 48 (17.0 mg, 88%) as a colorless solid. ESI–MS m/z 691 (M+H)+ as C34H50N4O7S2.
Methyl (7S)-6-N-((2′ S )-5′- n -propylazepane-2-carbonyl)-7-deoxy-7-(4-(pyrimidin-5-yl)phenylthio)-α-thiolincosaminide (49) (stereochemistry at the C-5′ position is not assigned)
Compound 48 (15.0 mg, 0.0217 mmol) and 2,2,2-trifluoroacetic acid (0.3 ml) were treated at 0 °C for 20 min and then treated room temperature for 1 h according to the similar procedure as described for the preparation of 44 to afford 49 (10.0 mg, 78%) as a colorless solid. [α]D24 +82.9° (c 0.84, MeOH); ESI–MS m/z 591 (M+H)+ as C29H42N4O5S2; TOF–ESI–HR-MS (M+H)+ calcd for C29H42N4O5S2: 591.2675, found: 591.2667; 1H NMR (400 MHz, CD3OD) δ 0.91 (t, J=7.1 Hz, 3 H), 1.21–1.45 (m, 5 H), 1.37 (d, J=6.8 Hz, 3 H), 1.45–1.55 (m, 1 H), 1.55–1.68 (m, 1 H), 1.80–1.91 (m, 1 H), 1.92–2.01 (m, 1 H), 1.94 (s, 3 H), 2.07–2.22 (m, 2 H), 2.98–3.08 (m, 1 H), 3.32–3.39 (m, 1 H), 3.60 (dd, J=10.2, 3.3 Hz, 1 H), 3.90 (dd, J=3.3, 0.8 Hz, 1 H), 3.95 (dq, J=6.8, 2.4 Hz, 1 H), 3.98 (dd, J=6.4, 5.1 Hz, 1 H), 4.10 (dd, J=10.2, 5.6 Hz, 1 H), 4.46 (dd, J=10.0, 0.8 Hz, 1 H), 4.64 (dd, J=10.0, 2.4 Hz, 1 H), 5.29 (d, J=5.6 Hz, 1 H), 7.52–7.59 (m, 2 H), 7.65–7.74 (m, 2 H), 9.07 (s, 2 H), 9.12 (s, 1 H).
Methyl 6-N-(2,2,2-trifluoroacetyl)-2,3,4,7-tetrakis-O-(trimethylsilyl)-α-thiolincosaminide (51)
Compound 50 (6.88 g, 19.7 mmol), trimethylchlorosilane (12.6 ml, 98.5 mmol) and hexamethyldisilazane (20.6 ml, 98.5 mmol) in pyridine (40 ml) were treated at room temperature for 1 h according to the similar procedure as described for the preparation of 22 to afford 51 (8.87 g, 71%) as a colorless solid. FAB–MS m/z 638 (M+H)+ as C23H50F3NO6SSi4.
Methyl 6-N-(2,2,2-trifluoroacetyl)-2,3,4-tris-O-(trimethylsilyl)-α-thiolincosaminide (52)
To a solution of compound 51 (8.87 g, 13.9 mmol) in MeOH (65 ml) was added 6 N acetic acid (4.17 ml) and stirred at room temperature for 15 min. The mixture was added to saturated aqueous NaHCO3 and concentrated under reduced pressure to remove MeOH. The desired compound was extracted with ethyl acetate, and then the organic phase was dried over Na2SO4, filtrated and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane/ethyl acetate=95/5 to 80/20) to obtain the title compound (7.21 g, 91% in 2 steps) as a colorless solid. ESI–MS m/z 566 (M+H)+ as C20H42F3NO6SSi3; 1H NMR (400 MHz, CD3OD) δ 0.133 (s, 9 H), 0.134 (s, 9 H), 0.14 (s, 9 H), 1.13 (d, J=6.5 Hz, 3 H), 2.06 (s, 3 H), 3.66 (dd, J=9.6, 2.4 Hz, 1 H), 3.92 (d, J=2.4 Hz, 1 H), 4.04 (dq, J=6.5, 4.5 Hz, 1 H), 4.14 (dd, J=9.6, 5.5 Hz, 1 H), 4.19 (d, J=9.6 Hz, 1 H), 4.40 (dd, J=9.6, 4.5 Hz, 1 H), 5.21 (d, J=5.5 Hz, 1 H).
Methyl (7S)-7-acetylthio-7-deoxy-6-N-(2,2,2-trifluoroacetyl)-2,3,4-tris-O-(trimethylsilyl)-α-thiolincosaminide (53)
Compound 52 (4.42 g, 7.82 mmol), Et3N (21.8 ml, 15.6 mmol) and methanesulfonyl chloride (1.21 ml, 15.6 mmol) in CHCl3 (20 ml) were treated at room temperature for 1 h according to the similar procedure as described for the preparation of 26 to afford methyl 7-O-methaneslufonyl-6-N-(2,2,2-trifluoroacetyl)-2,3,4-tris-O-(trimethylsilyl)-α-thiolincosaminide (5.46 g, quant) as a colorless solid. To a solution of this mesylate (5.46 g, 7.82 mmol) in DMF (40 ml) was added AcSK (2.68 g, 23.4 mmol) and stirred at 80 °C for 1.5 h. The mixture was concentrated under reduced pressure, diluted with ethyl acetate and saturated aqueous NaHCO3, and extracted with ethyl acetate. The organic phase was dried over Na2SO4, filtrated and concentrated under reduced pressure. To the resulting residue were added pyridine (16 ml), trimethylchlorosilane (6.35 ml, 50.0 mmol) and hexamethyldisilazane (10.5 ml, 50.0 mmol), and stirred at room temperature for 3 h. The mixture was added to saturated aqueous NaHCO3, extracted with ethyl acetate and then the organic phase was dried over Na2SO4, filtrated and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane to hexane/ethyl acetate=10/1) to obtain the title compound (2.88 g) as a crude compound. The crude compound (306.3 mg) was suspended in hexane, and then the solid was filtered, washed with hexane to obtain the title compound (129 mg, 25% in 3 steps) as a colorless solid. As compound 53 is partially soluble in hexane, a yield of the third step was low. ESI–MS m/z 624 (M+H)+ as C22H44F3NO6S2Si3; 1H NMR (400 MHz, CDCl3) δ 0.15 (s, 9 H), 0.16 (s, 9 H), 0.22 (s, 9 H), 1.41 (d, J=7.1 Hz, 3 H), 2.01 (s, 3 H), 2.33 (s, 3 H), 3.65 (dd, J=9.5, 2.7 Hz, 1 H), 3.76–3.86 (m, 1 H), 3.97–4.04 (m, 1 H), 4.10 (dd, J=9.5, 5.4 Hz, 1 H), 4.14–4.21 (m, 1 H), 4.34-4.44 (m, 1 H), 5.18 (d, J=5.4 Hz, 1 H), 7.20 (br dd, J=9.5 Hz, 1 H).
Methyl (7S)-7-deoxy-7-mercapto-6-N-(2,2,2-trifluoroacetyl)-α-thiolincosaminide (54)
Compound 53 (2.83 g, 4.54 mmol) and 1 N HCl (18.1 ml) in MeOH (30 ml) were treated at room temperature for 10 min according to the similar procedure as described for the preparation of 30, and then the crude intermediate and 28% NaOMe/MeOH solution (2.63 ml, 8.68 mmol) in MeOH (25 ml) at room temperature were treated for 15 min according to the similar procedure as described for the preparation of 30 to afford 54 (1.65 g, 99% in 2 steps from 53) as a colorless solid. ESI–MS m/z 366 (M+H)+ as C11H18F3NO5S2; 1H NMR (400 MHz, CD3OD) δ 1.29 (d, J=7.0 Hz, 3 H), 2.16 (s, 3 H), 3.45 (dq, J=7.0, 2.2 Hz, 1 H), 3.54 (dd, J=10.2, 3.2 Hz, 1 H), 3.82 (dd, J=3.2, 1.0 Hz, 1 H), 4.08 (dd, J=10.2, 5.6 Hz, 1 H), 4.39 (dd, J=9.9, 1.0 Hz, 1 H), 4.55 (dd, J=9.9, 2.2 Hz, 1 H), 5.26 (d, J=5.6 Hz, 1 H).
Methyl (7S)-7-deoxy-7-((4-(2-dimethylaminoethyl)phenyl)thio-6-N-(2,2,2-trifluoroacetyl)-α-thiolincosaminide (55)
Compound 54 (3.44 g, 9.42 mmol), 2-(4-bromophenyl)-N,N-dimethylethanamine (3.22 g, 14.1 mmol), Xantphos (544.8 mg, 0.942 mmol), Pd2(dba)3 (431.1 mg, 0.471 mmol) and N,N-diisopropylethylamine (3.28 ml, 18.8 mmol) in 1,4-dioxane (37 ml) were treated for 17 h according to the similar procedure as described for the preparation of 40 to afford 55 (3.85 g, 80%) as a colorless solid. FAB–MS m/z 513 (M+H)+ as C21H31F3N2O5S2; 1H NMR (400 MHz, CD3OD) δ 1.27 (d, J=6.9 Hz, 3 H), 2.01 (s, 3 H), 2.36 (s, 6 H), 2.58-2.68 (m, 2 H), 2.74-2.84 (m, 2 H), 3.59 (dd, J=10.3, 3.2 Hz, 1 H), 3.76 (dq, J=6.9, 2.8 Hz, 1 H), 3.87-3.92 (m, 1 H), 4.09 (dd, J=10.3, 5.6 Hz, 1 H), 4.59 (dd, J=9.4, 0.7 Hz, 1 H), 4.64 (dd, J=9.4, 2.8 Hz, 1 H), 5.28 (d, J=5.6 Hz, 1 H), 7.16–7.22 (m, 2 H), 7.33-7.39 (m, 2 H).
Methyl (7S)-7-deoxy-7-((4-(2-dimethylaminoethyl)phenyl)thio-α-thiolincosaminide (56)
To a solution of 55 (3.85 g, 7.51 mmol) in CH2Cl2 (73 ml) were added N-benzyl-N, N, N-triethylammonium bromide (171.1 mg, 0.751 mmol) and 20% aqueous potassium hydroxide (5.1 ml), stirred at room temperature for 4 h. To a solution of mixture was added 1 N HCl to adjust at pH 7 and then the solution was concentrated under reduced pressure. The resulting residue was diluted with MeOH, filtrated, and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (CHCl3/MeOH/28% aq NH4OH=20/1/0.1 to 10/1/0.1) to obtain the title compound (2.94 g, 94%) as off white solid. 1H NMR (400 MHz, CD3OD) δ 1.41 (d, J=7.1 Hz, 3 H), 1.92 (s, 3 H), 2.30 (s, 6 H), 2.51–2.59 (m, 2 H), 2.73–2.81 (m, 2 H), 3.20 (dd, J=8.8, 2.7 Hz, 1 H), 3.59 (dd, J=10.3, 3.4 Hz, 1 H), 3.63 (dq, J=7.1, 2.7 Hz, 1 H), 4.04–4.13 (m, 2 H), 4.23 (dd, J=8.8, 1.2 Hz, 1 H), 5.22 (d, J=5.8 Hz, 1 H), 7.14–7.20 (m, 2 H), 7.31–7.37 (m, 2 H).
Methyl (7S)-6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(cyclopropylmethyl)piperidine-2′-carbonyl)-7-deoxy-7-((4-(2-dimethylaminoethyl)phenyl)thio-α-thiolincosaminide (57)
Compound 15 (1.15 g, 4.04 mmol), 56 (2.02 g, 4.85 mmol), 1-hydroxybenzotriazole (0.819 g, 6.06 mmol) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (1.16 g, 6.06 mmol) in DMF (20 ml) were treated for 5.5 h according to the similar procedure as described for the preparation of 34 to afford the title compound (2.00 g, 73%) as a colorless solid. ESI–MS m/z 682 (M+H)+ as C34H55N3O7S2.
Methyl (7S)-6-N-((2′S, 4′R)-4′-(cyclopropylmethyl)piperidine-2′-carbonyl)-7-deoxy-7-((4-(2-dimethylaminoethyl)phenyl)thio-α-thiolincosaminide (58)
Compound 57 (2.00 g, 2.93 mmol) and 2,2,2-trifluoroacetic acid (3.25 ml) in CH2Cl2 (5.0 ml) were treated at 0 °C for 3.5 h according to the similar procedure as described for the preparation of 44 to afford 58 (1.69 g, 99%) as a colorless solid. [α]D23 +101.3° (c 0.65, MeOH); ESI–MS m/z 582 (M+H)+ as C29H47N3O5S2; TOF–ESI–HR-MS (M+H)+ calcd for C29H47N3O5S2: 582.3035, found: 582.3032; 1H NMR (400 MHz, CD3OD) δ −0.02 to 0.06 (m, 2 H), 0.40–0.50 (m, 2 H), 0.67–0.78 (m, 1 H), 1.02–1.25 (m, 4 H), 1.25 (d, J=7.0 Hz, 3 H), 1.60–1.74 (m, 1 H), 1.75–1.84 (m, 1 H), 1.97 (s, 3 H), 2.04–2.12 (m, 1 H), 2.31 (s, 6 H), 2.52–2.61 (m, 2 H), 2.62–2.72 (m, 1 H), 2.73–2.81 (m, 2 H), 3.13–3.21 (m, 1 H), 3.34 (dd, J=11.8, 2.9 Hz, 1 H), 3.56 (dd, J=10.3, 3.3 Hz, 1 H), 3.76 (dq, J=7.0, 2.4 Hz, 1 H), 3.85 (dd, J=3.3, 0.8 Hz, 1 H), 4.08 (dd, J=10.3, 5.6 Hz, 1 H), 4.40 (dd, J=9.9, 0.8 Hz, 1 H), 4.52 (dd, J=9.9, 2.4 Hz, 1 H), 5.25 (d, J=5.6 Hz, 1 H), 7.15–7.19 (m, 2 H), 7.29–7.41 (m, 2 H).
Methyl (7S)-6-N-((2′S, 4′R)-4′-(cyclopropylmethyl)-1′-N-methylpiperidine-2′-carbonyl)-7-deoxy-7-((4-(2-dimethylaminoethyl)phenyl)thio-α-thiolincosaminide (59)
To a solution of 58 (1.19 g, 2.04 mmol) in MeOH (21 ml) were added 36% aqueous HCHO (0.51 ml, 6.12 mmol), AcOH (0.35 ml, 6.12 mmol) and NaBH(OAc)3 (2.59 g, 12.2 mmol), and stirred at room temperature for 1 h. The mixture was diluted with ethyl acetate and saturated aqueous NaHCO3, extracted with ethyl acetate/MeOH=5/1. The organic phase was dried over Na2SO4, filtrated and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (CHCl3/MeOH/28% aq NH4OH=20/1/0.1) to obtain the title compound (1.10 g, 91%) as a colorless solid. [α]D22 +88.4° (c 1.58, MeOH); ESI–MS m/z 596 (M+H)+ as C30H49N3O5S2; TOF–ESI–HR-MS (M+H)+ calcd for C30H49N3O5S2: 596.3192, found: 596.3188; 1H NMR (400 MHz, CD3OD) δ −0.05 to 0.06 (m, 2 H), 0.38–0.49 (m, 2 H), 0.63–0.76 (m, 1 H), 1.13–1.38 (m, 4 H), 1.27 (d, J=7.0 Hz, 3 H), 1.40–1.57 (m, 1 H), 1.75–1.84 (m, 1 H), 1.92–2.10 (m, 1 H), 1.99 (s, 3 H), 2.10–2.21 (m, 1 H), 2.26 (s, 3 H), 2.41 (s, 6 H), 2.60–2.73 (m, 3 H), 2.77–2.86 (m, 2 H), 2.92–3.02 (m, 1 H), 3.58 (dd, J=10.1, 3.2 Hz, 1 H), 3.75–3.85 (m, 2 H), 4.10 (dd, J=10.1, 5.6 Hz, 1 H), 4.41 (br dd, J=9.8, 0.6 Hz, 1 H), 4.54 (dd, J=9.8, 2.6 Hz, 1 H), 5.27 (d, J=5.6 Hz, 1 H), 7.16–7.23 (m, 2 H), 7.33–7.39 (m, 2 H).
Methyl (7S)-6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(cyclopropylmethyl)piperidine-2′-carbonyl)-7-deoxy-7-(4-(pyrrolidin-1-ylmethyl)phenylthio)-α-thiolincosaminide (60)
Compound 33 (40.2 mg, 0.0739 mmol), 1-(4-bromobenzyl)pyrrolidine (64.6 mg, 0.269 mmol), Xantphos (4.6 mg, 0.0077 mmol), Pd2(dba)3 (4.0 mg, 4.3 μmol) and N,N-diisopropylethylamine (38.5 μl, 0.22 mmol) in 1,4-dioxane (1 ml) were treated for 4 h according to the similar procedure as described for the preparation of 40 to afford 60 (41.4 mg, 81%) as a colorless solid. FAB–MS m/z 694 (M+H)+ as C35H55N3O7S2.
Methyl (7S)-6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(cyclopropylmethyl)piperidine-2′-carbonyl)-7-deoxy-7-(4-(1-methyl-1,2,5,6-tetrahydropyridin-3-yl)phenylthio)-α-thiolincosaminide (61)
Compound 33 (222.4 mg, 0.416 mmol), 3-(4-bromophenyl)-1-methyl-1,2,5,6-tetrahydropyridine (125.6 mg, 0.498 mmol), Xantphos (25.8 mg, 0.043 mmol), Pd2(dba)3 (19.4 mg, 0.021 mmol) and N,N-diisopropylethylamine (144 μl, 0.83 mmol) in 1,4-dioxane (3.5 ml) were treated for 5 h according to the similar procedure as described for the preparation of 40 to afford 61 (251.9 mg, 86%) as a colorless solid. ESI–MS m/z 706 (M+H)+ as C36H55N3O7S2.
Methyl (7S)-6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(cyclopropylmethyl)piperidine-2′-carbonyl)-7-deoxy-7-(4-(1-methylpiperidin-3-yl)phenylthio)-α-thiolincosaminide (62) (a diastereo mixture at an N-methylpiperidine ring)
To a solution of 61 (201.7 mg, 0.286 mmol) in toluene (10 ml) was added 4-methylbenzenesulfonohydrazide (1.10 g, 5.72 mmol) at room temperature and then refluxed for 3 h. To the mixture was further added 4-methylbenzenesulfonohydrazide (1.09 g, 5.70 mmol) and then stirred for 2.5 h under the reflux condition. The solution was added to 1 N NaOH, extracted with ethyl acetate. The organic phase was dried over Na2SO4, filtrated and concentrated under reduced pressure. The resulting residue was purified by preparative TLC (CHCl3/MeOH/28% aq NH4OH=10/1/0.1) to obtain the title compound (27.7 mg, 14%) as a colorless solid. ESI–MS m/z 708 (M+H)+ as C36H57N3O7S2.
Methyl (7S)-6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(cyclopropylmethyl)piperidine-2′-carbonyl)-7-deoxy-7-(4-(pyridin-3-yl)phenylthio)-α-thiolincosaminide (63)
Compound 33 (30.0 mg, 0.0561 mmol), 3-(4-iodophenyl)pyridine (18.9 mg, 0.0673 mmol), Xantphos (7.8 mg, 0.0135 mmol), Pd2(dba)3 (5.1 mg, 5.6 μmol) and N,N-diisopropylethylamine (19.5 μl, 0.112 mmol) in 1,4-dioxane (0.5 ml) were treated for 5 h according to the similar procedure as described for the preparation of 40 to afford 63 (39.3 mg as crude).
Methyl (7S)-6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(cyclopropylmethyl)piperidine-2′-carbonyl)-7-deoxy-7-(4-(pyrazin-2-yl)phenylthio)-α-thiolincosaminide (64)
Compound 33 (80 mg, 0.147 mmol), 2-(4-bromophenyl)pyrazine (50.0 mg, 0.213 mmol), Xantphos (10 mg, 0.0173 mmol), Pd2(dba)3 (10 mg, 0.0109 mmol) and N,N-diisopropylethylamine (60 μl, 0.35 mmol) in 1,4-dioxane (1.5 ml) were treated for 4 h according to the similar procedure as described for the preparation of 40 to afford 64 as a crude compound.
Methyl (7S)-6-N-((2′S, 4′R)-1′-N-(tert-butoxycarbonyl)-4′-(cyclopropylmethyl)piperidine-2′-carbonyl)-7-deoxy-7-(4-(1,2,3-thiadiazol-4-yl)phenylthio)-α-thiolincosaminide (65)
Compound 33 (80.0 mg, 0.147 mmol), 4-(4-bromophenyl)-1,2,3-thiadiazole (50 mg, 0.207 mmol), Xantphos (10 mg, 0.0173 mmol), Pd2(dba)3 (10 mg, 0.011 mmol) and N,N-diisopropylethylamine (60 μl, 0.35 mmol) in 1,4-dioxane (1.5 ml) were treated for 4 h according to the similar procedure as described for the preparation of 40 to afford 65 as a crude compound. ESI–MS m/z 695 (M+H)+ as C32H46N4O7S3.
Methyl (7S)-6-N-((2′S, 4′R)-4′-(cyclopropylmethyl)piperidine-2′-carbonyl)-7-deoxy-7-(4-(pyrrolidin-1-ylmethyl)phenylthio)-α-thiolincosaminide (66)
Compound 60 (41.4 mg, 0.0597 mmol) and 2,2,2-trifluoroacetic acid (0.09 ml) in CH2Cl2 (0.9 ml) were treated at −20 °C for 5 min and then treated at room temperature for 5 h according to the similar procedure as described for the preparation of 44 to afford 66 (28.8 mg, 81%) as a colorless solid. [α]D23 +68.9° (c 0.20, MeOH); ESI–MS m/z 594 (M+H)+ as C30H47N3O5S2; TOF–ESI–HR-MS (M+H)+ calcd for C30H47N3O5S2: 594.3035, found: 594.3031; 1H NMR (400 MHz, CD3OD) δ −0.01 to 0.06 (m, 2 H), 0.39–0.50 (m, 2 H), 0.66–0.79 (m, 1 H), 1.02-1.26 (m, 4 H), 1.28 (d, J=6.8 Hz, 3 H), 1.60–1.74 (m, 1 H), 1.74–1.87 (m, 5 H), 1.93 (s, 3 H), 2.05–2.17 (m, 1 H), 2.49–2.60 (m, 4 H), 2.62–2.74 (m, 1 H), 3.12–3.24 (m, 1 H), 3.37 (dd, J=11.8, 2.8 Hz, 1 H), 3.57 (dd, J=10.2, 3.3Hz, 1 H), 3.63 (s, 2 H), 3.81 (dq, J=6.8, 2.3 Hz, 1 H), 3.86 (br dd, J=3.3, 0.7 Hz, 1 H), 4.08 (dd, J=10.2, 5.6 Hz, 1 H), 4.38–4.46 (m, 1 H), 4.55 (dd, J=10.1, 2.3 Hz, 1 H), 5.26 (d, J=5.6 Hz, 1 H), 7.26–7.32 (m, 2 H), 7.34–7.39 (m, 2 H).
Methyl (7S)-6-N-((2′S, 4′R)-4′-(cyclopropylmethyl)-1′-N-methylpiperidine-2′-carbonyl)-7-deoxy-7-(4-(pyrrolidin-1-ylmethyl)phenylthio)-α-thiolincosaminide (67)
Compound 66 (16.4 mg, 0.0276 mmol), 36% aqueous formaldehyde (21 μl, 0.28 mmol), AcOH (16 μl, 0.28 mmol) and NaBH(OAc)3 (61.4 mg, 0.281 mmol) in MeOH (1.0 ml) were treated at room temperature for 2 h according to the similar procedure as described for the preparation of 59 to afford 67 (14.7 mg, 88%) as a colorless solid. [α]D22 +65.9° (c 0.11, MeOH); ESI–MS m/z 608 (M+H)+ as C31H49N3O5S2; TOF–ESI–HR-MS (M+H)+ calcd for C31H49N3O5S2: 608.3192, found: 608.3187; 1H NMR (400 MHz, CD3OD) δ −0.05 to 0.07 (m, 2 H), 0.37–0.51 (m, 2 H), 0.64–0.78 (m, 1 H), 1.10–1.22 (m, 2 H), 1.23–1.39 (m, 2 H), 1.30 (d, J=6.8 Hz, 3 H), 1.41–1.58 (m, 1 H), 1.73–1.88 (m, 5 H), 1.91–2.01 (m, 1 H), 1.95 (s, 3 H), 2.07–2.19 (m, 1 H), 2.56 (s, 3 H), 2.53–2.66 (m, 5 H), 2.92–3.01 (m, 1 H), 3.57 (dd, J=10.2, 3.2 Hz, 1 H), 3.65 (s, 2 H), 3.79–3.89 (m, 2 H), 4.10 (dd, J=10.2, 5.6 Hz, 1 H), 4.37–4.44 (m, 1 H), 4.56 (dd, J=9.9, 2.6 Hz, 1 H), 5.26 (d, J=5.6 Hz, 1 H), 7.26-7.33 (m, 2 H), 7.35–7.43 (m, 2 H).
Methyl (7S)-6-N-((2′S, 4′R)-4′-(cyclopropylmethyl)piperidine-2′-carbonyl)-7-deoxy-7-(4-(1-methyl-1,2,5,6-tetrahydropyridin-3-yl)phenylthio)-α-thiolincosaminide (68)
Compound 61 (26.6 mg, 0.37 mmol) and 2,2,2-trifluoroacetic acid (60 μl) in CH2Cl2 (0.6 ml) were treated at −20 °C for 20 min and then treated room temperature for 4 h according to the similar procedure as described for the preparation of 44 to afford 68 (21.5 mg, 94%) as a colorless solid. [α]D22 +91.4° (c 1.82, MeOH); ESI–MS m/z 606 (M+H)+ as C31H47N3O5S2; TOF–ESI–HR-MS (M+H)+ calcd for C31H47N3O5S2: 606.3035, found: 606.3012; 1H NMR (400 MHz, CD3OD) δ −0.07–0.03 (m, 2 H), 0.38–0.48 (m, 2 H), 0.62–0.75 (m, 1 H), 1.07–1.25 (m, 4 H), 1.25 (d, J=6.8 Hz, 3 H), 1.63–1.78 (m, 1 H), 1.78–1.87 (m, 1 H), 1.90 (s, 3 H), 2.10–2.19 (m, 1 H), 2.33–2.41 (m, 2 H), 2.44 (s, 3 H), 2.66 (t, J=5.9 Hz, 2 H), 2.80 (dt, J=12.9, 2.7 Hz, 1 H), 3.21–3.39 (m, 1 H), 3.32–3.38 (m, 2 H), 3.53–3.62 (m, 2 H), 3.80 (dq, J=6.8, 2.4 Hz, 1 H), 3.89 (dd, J=3.2, 0.7 Hz, 1 H), 4.09 (dd, J=10.3, 5.6 Hz, 1 H), 4.43 (dd, J=10.0, 0.7 Hz, 1 H), 4.57 (dd, J=10.0, 2.4 Hz, 1 H), 5.27 (d, J=5.6 Hz, 1 H), 6.17–6.23 (m, 1 H), 7.29–7.40 (m, 4 H).
Methyl (7S)-6-N-((2′S, 4′R)-4′-(cyclopropylmethyl)-1′-N-methylpiperidine-2′-carbonyl)-7-deoxy-7-(4-(1-methyl-1,2,5,6-tetrahydropyridin-3-yl)phenylthio)-α-thiolincosaminide (69)
Compound 68 (38.4 mg, 0.0634 mmol), 36% aqueous formaldehyde (48.0 μl, 0.645 mmol), AcOH (36.5 μl, 0.638 mmol) and NaBH(OAc)3 (141.2 mg, 0.633 mmol) in MeOH (2.2 ml) were treated at room temperature for 1 h according to the similar procedure as described for the preparation of 59 to afford 69 (36.0 mg, 92%) as an off white solid. [α]D23 +67.4° (c 2.48, MeOH); ESI–MS m/z 620 (M+H)+ as C32H49N3O5S2; TOF–ESI–HR-MS (M+H)+ calcd for C32H49N3O5S2: 620.3192, found: 620.3187; 1H NMR (400 MHz, CD3OD) δ −0.05 to 0.06 (m, 2 H), 0.38-0.49 (m, 2 H), 0.63–0.76 (m, 1 H), 1.10–1.23 (m, 2 H), 1.24–1.43 (m, 2 H), 1.30 (d, J=7.0 Hz, 3 H), 1.52–1.67 (m, 1 H), 1.81–1.90 (m, 1 H), 1.94 (s, 3 H), 2.00–2.09 (m, 1 H), 2.31–2.46 (m, 1 H), 2.41 (s, 3 H), 2.46–2.56 (m, 2 H), 2.69 (s, 3 H), 2.90–3.00 (m, 3 H), 3.06–3.15 (m, 1 H), 3.59 (dd, J=10.2, 3.2 Hz, 1 H), 3.62–3.69 (m, 2 H), 3.78–3.87 (m, 2 H), 4.10 (dd, J=10.2, 5.6 Hz, 1 H), 4.43 (dd, J=9.9, 0.6 Hz, 1 H), 4.59 (dd, J=9.9, 2.6 Hz, 1 H), 5.26 (d, J=5.6 Hz, 1 H), 6.23–6.29 (m, 1 H), 7.32–7.42 (m, 4 H).
Methyl (7S)-6-N-((2′S, 4′R)-4′-(cyclopropylmethyl)piperidine-2′-carbonyl)-7-deoxy-7-(4-(1-methylpiperidin-3-yl)phenylthio)-α-thiolincosaminide (70) (a diastero mixture at an N-methylpiperidine ring)
Compound 62 (6.9 mg, 9.75 μmol) and 2,2,2-trifluoroacetic acid (20 μl) in CH2Cl2 (0.2 ml) were treated at −20 °C for 20 min, and then treated room temperature for 4 h according to the similar procedure as described for the preparation of 44 to afford 70 (4.5 mg, 76%) as a colorless solid. [α]D23 +81.6° (c 0.65, MeOH); ESI–MS m/z 608 (M+H)+ as C31H49N3O5S2; TOF–ESI–HR-MS (M+H)+ calcd for C31H49N3O5S2: 608.3192, found: 608.3175; 1H NMR (400 MHz,CD3OD) δ −0.06–0.04 (m, 2 H), 0.37–0.47 (m, 2 H), 0.60–0.75 (m, 1 H), 1.08–1.28 (m, 4 H), 1.22 (d, J=6.9 Hz, 3 H), 1.38–1.52 (m, 1 H), 1.63–1.78 (m, 2 H), 1.78–1.89 (m, 3 H), 1.90 (s, 3 H), 2.12–2.24 (m, 3 H), 2.37 (s, 3 H), 2.72–2.83 (m, 2 H), 2.94–3.04 (m, 2 H), 3.18–3.26 (m, 1 H), 3.46–3.56 (m, 2 H), 3.73 (dq, J=6.9, 2.3 Hz, 1 H), 3.79–3.85 (m, 1 H), 4.03 (dd, J=10.2, 5.6 Hz, 1 H), 4.37 (m, 1 H), 4.50 (dd, J=9.9, 2.3 Hz, 1 H), 5.21 (d, J=5.6 Hz, 1 H), 7.12-7.19 (m, 2 H), 7.28-7.34 (m, 2 H).
Methyl (7S)-6-N-((2′S, 4′R)-4′-(cyclopropylmethyl)-1’-N-methylpiperidine-2′-carbonyl)-7-deoxy-7-(4-(1-methylpiperidin-3-yl)phenylthio)-α-thiolincosaminide (71) (a diastero mixture at an N-methylpiperidine ring)
Compound 70 (9.4 mg, 0.016 mmol), 36% aqueous formaldehyde (12 μl, 0.16 mmol), AcOH (9.0 μl, 0.16 mmol) and NaBH(OAc)3 (37.1 mg, 0.166 mmol) in MeOH (0.6 ml) were treated at room temperature for 1.5 h according to the similar procedure as described for the preparation of 59 to afford 71 (9.4 mg, 98%) as a colorless solid. [α]D22 +70.1° (c 0.15, MeOH); ESI–MS m/z 622 (M+H)+ as C32H51N3O5S2; TOF–ESI–HR-MS (M+H)+ calcd for C32H51N3O5S2: 622.3348, found: 622.3342; 1H NMR (400 MHz, CD3OD) δ −0.28 to −0.16 (m, 2 H), 0.29–0.39 (m, 2 H), 0.55–0.67 (m, 1 H), 1.04–1.14 (m, 2 H), 1.15–1.31 (m, 2 H), 1.19 (d, J=6.8 Hz, 3 H), 1.38–1.57 (m, 2 H), 1.65–1.78 (m, 2 H), 1.79–1.89 (m, 2 H), 1.86 (s, 3 H), 1.89–1.97 (m, 1 H), 2.09–2.20 (m, 1 H), 2.23 (s, 3 H), 2.32–2.45 (m, 2 H), 2.47 (s, 3 H), 2.65 (br dd, J=11.5, 2.4 Hz, 1 H), 2.73–2.84 (m, 1 H), 2.90–2.97 (m, 1 H), 3.05–3.15 (m, 2 H), 3.48 (dd, J=10.2, 3.2 Hz, 1 H), 3.64–3.76 (m, 2 H), 4.00 (dd, J=10.2, 5.6 Hz, 1 H), 4.30 (d, J=9.8 Hz, 1 H), 4.46 (dd, J=9.8, 2.6 Hz, 1 H), 5.16 (d, J=5.6 Hz, 1 H), 7.09-7.16 (m, 2 H), 7.25-7.34 (m, 2 H).
Methyl (7S)-6-N-((2′S, 4′R)-4′-(cyclopropylmethyl)piperidine-2′-carbonyl)-7-deoxy-7-(4-(pyridin-3-yl)phenylthio)-α-thiolincosaminide (72)
Compound 63 (39.3 mg, 0.0571 mmol) and 2,2,2-trifluoroacetic acid (0.5 ml) in CH2Cl2 (0.1 ml) were treated at 0 °C for 1 h according to the similar procedure as described for the preparation of 44 to afford 72 (17.2 mg, 52% in 2 steps from 33) as a colorless solid. [α]D23 +92.4° (c 1.12, MeOH); ESI–MS m/z 588 (M+H)+ as C30H41N3O5S2; TOF–ESI–HR-MS (M+H)+ calcd for C30H41N3O5S2: 588.2566, found: 588.2560; 1H NMR (400 MHz, CD3OD) δ −0.04 to 0.05 (m, 2 H), 0.39–0.49 (m, 2 H), 0.65–0.76 (m, 1 H), 1.09–1.25 (m, 4 H), 1.35 (d, J=7.0 Hz, 3 H), 1.65–1.79 (m, 1 H), 1.79–1.88 (m, 1 H), 1.95 (s, 3 H), 2.10–2.20 (m, 1 H), 3.23 (dt, J=10.9, 1.9 Hz, 1 H), 3.18–3.27 (m, 1 H), 3.49 (dd, J=12.0, 2.9 Hz, 1 H), 3.59 (dd, J=10.2, 3.4 Hz, 1 H), 3.86–3.95 (m, 2 H), 4.10 (dd, J=10.2, 5.6 Hz, 1 H), 4.45 (br dd, J=9.9, 0.7 Hz, 1 H), 4.61 (dd, J=9.9, 2.4 Hz, 1 H), 5.28 (d, J=5.6 Hz, 1 H), 7.46-7.55 (m, 3 H), 7.58-7.66 (m, 2 H), 8.07 (ddd, J=7.9, 2.3, 1.5 Hz, 1 H), 8.50 (dd, J=4.9, 1.5 Hz, 1 H), 8.79 (dd, J=2.3, 0.8 Hz, 1 H).
Methyl (7S)-6-N-((2′S, 4′R)-4′-(cyclopropylmethyl)-1′-N-methylpiperidine-2′-carbonyl)-7-deoxy-7-(4-(pyridin-3-yl)phenylthio)-α-thiolincosaminide (73)
Compound 72 (20.0 mg, 0.034 mmol), 36% aqueous formaldehyde (25 μl, 0.34 mmol), AcOH (19 μl, 0.30 mmol) and NaBH(OAc)3 (18 mg, 0.08 mmol) in MeOH (2.2 ml) were treated at 0 °C for 1 h according to the similar procedure as described for the preparation of 59 to afford 73 (20.0 mg, 98%) as a colorless solid. [α]D24 +57.4° (c 0.28, MeOH); ESI–MS m/z 602 (M+H)+ as C31H43N3O5S2; TOF–ESI–HR-MS (M+H)+ calcd for C31H43N3O5S2: 602.2722, found: 602.2714; 1H NMR (400 MHz, CD3OD) δ −0.05 to 0.05 (m, 2 H), 0.38–0.48 (m, 2 H), 0.62–0.74 (m, 1 H), 1.07–1.43 (m, 4 H), 1.37 (d, J=7.0 Hz, 3 H), 1.54–1.69 (m, 1 H), 1.83–1.92 (m, 1 H), 1.96 (s, 3 H), 2.03–2.11 (m, 1 H), 2.40–2.55 (m, 1 H), 2.47 (s, 3 H), 2.98–3.08 (m, 1 H), 3.10–3.20 (m, 1 H), 3.59 (dd, J=10.3, 3.3 Hz, 1 H), 3.85 (br dd, J=3.2, 0.9 Hz, 1 H), 3.91 (dq, J=6.9, 2.6 Hz, 1 H), 4.10 (dd, J=10.2, 5.6 Hz, 1 H), 4.46 (br dd, J=9.9, 0.7 Hz, 1 H), 4.64 (dd, J=9.9, 2.6 Hz, 1 H), 5.27 (d, J=5.5 Hz, 1 H), 7.50–7.56 (m, 2 H), 7.51 (ddd, J=8.0, 4.9, 0.8 Hz, 1 H), 7.60–7.67 (m, 2 H), 8.08 (ddd, J=8.0, 2.3, 1.5 Hz, 1 H), 8.51 (dd, J=4.9, 1.5 Hz, 1 H), 8.79 (br dd, J=2.3, 0.8 Hz, 1 H).
Methyl (7S)-6-N-((2′S, 4′R)-4′-(cyclopropylmethyl)piperidine-2′-carbonyl)-7-deoxy-7-(4-(pyrazin-2-yl)phenylthio)-α-thiolincosaminide (74)
Compound 64 (crude) and 2,2,2-trifluoroacetic acid (0.5 ml) were treated at 0 °C for 30 min according to the similar procedure as described for the preparation of 44 to afford 74 (41.1 mg, 48% in 2 steps from 33) as a colorless solid. [α]D22 +78.5° (c 1.20, MeOH); ESI–MS m/z 589 (M+H)+ as C29H40N4O5S2; TOF–ESI–HR-MS (M+H)+ calcd for C29H40N4O5S2: 589.2518, found: 589.2514; 1H NMR (400 MHz, CD3OD) δ −0.03 to 0.04 (m, 2 H), 0.40–0.48 (m, 2 H), 0.64–0.75 (m, 1 H), 1.09–1.27 (m, 4 H), 1.38 (d, J=6.9 Hz, 3 H), 1.65–1.78 (m, 1 H), 1.79–1.87 (m, 1 H), 1.91 (s, 3 H), 2.10–2.18 (m, 1 H), 2.70–2.81 (m, 1 H), 3.19–3.26 (m, 1 H), 3.49 (dd, J=12.0, 2.9 Hz, 1 H), 3.59 (dd, J=10.2, 3.2 Hz, 1 H), 3.89 (dd, J=3.2, 0.8 Hz, 1 H), 3.94 (dq, J=6.9, 2.4 Hz, 1 H), 4.10 (dd, J=10.2, 5.6 Hz, 1 H), 4.45 (dd, J=10.0, 0.8 Hz, 1 H), 4.63 (dd, J=10.0, 2.4 Hz, 1 H), 5.28 (d, J=5.6 Hz, 1 H), 7.48-7.55 (m, 2 H), 7.99-8.06 (m, 2 H), 8.50 (d, J=2.6 Hz, 1 H), 8.64 (dd, J=2.6, 1.5 Hz, 1 H), 9.08 (d, J=1.5 Hz, 1 H).
Methyl (7S)-6-N-((2′S, 4′R)-4′-(cyclopropylmethyl)-1′-N-methylpiperidine-2′-carbonyl)-7-deoxy-7-(4-(pyrazin-2-yl)phenylthio)-α-thiolincosaminide (75)
Compound 74 (31.0 mg, 0.050 mmol), 36% aqueous formaldehyde (12 μl, 0.15 mmol), AcOH (17 μl, 0.30 mmol) and NaBH(OAc)3 (31.6 mg, 0.15 mmol) in MeOH (0.5 ml) were treated at room temperature for 30 min according to the similar procedure as described for the preparation of 59 to afford 75 (28.4 mg, 94%) as a colorless solid. [α]D25 +72.4° (c 0.64, MeOH); ESI–MS m/z 603 (M+H)+ as C30H42N4O5S2; TOF–ESI–HR-MS (M+H)+ calcd for C30H42N4O5S2: 603.2675, found: 603.2672; 1H NMR (400 MHz, CD3OD) δ −0.06 to 0.04 (m, 2 H), 0.37–0.47 (m, 2 H), 0.60–0.74 (m, 1 H), 1.08–1.22 (m, 2 H), 1.25–1.38 (m, 2 H), 1.39 (d, J=7.0 Hz, 3 H), 1.46–1.60 (m, 1 H), 1.78–1.85 (m, 1 H), 1.92 (s, 3 H), 1.95–2.02 (m, 1 H), 2.20–2.30 (m, 1 H), 2.34 (s, 3 H), 2.77 (br dd, J=11.4, 2.1 Hz, 1 H), 2.99–3.07 (m, 1 H), 3.59 (dd, J=10.2, 3.2 Hz, 1 H), 3.85 (dd, J=3.2, 0.6 Hz, 1 H), 3.96 (dq, J=6.9, 2.6 Hz, 1 H), 4.11 (dd, J=10.2, 5.6 Hz, 1 H), 4.45 (br dd, J=9.9, 0.6 Hz, 1 H), 4.64 (dd, J=9.9, 2.6 Hz, 1 H), 5.27 (d, J=5.6 Hz, 1 H), 7.50–7.56 (m, 2 H), 8.01–8.07 (m, 2 H), 8.50 (d, J=2.5 Hz, 1 H), 8.65 (dd, J=2.5, 1.5 Hz, 1 H), 9.09 (d, J=1.5 Hz, 1 H).
Methyl (7S)-6-N-((2′S, 4′R)-4′-(cyclopropylmethyl)-1′-N-methylpiperidine-2′-carbonyl)-7-deoxy-7-(4-(pyrimidin-5-yl)phenylthio)-α-thiolincosaminide (76)
Compound 47 (894.4 mg, 1.52 mmol), 36% aqueous formaldehyde (1.10 ml, 14.8 mmol), AcOH (0.85 ml, 14.8 mmol) and NaBH(OAc)3 (3.31 g, 14.8 mmol) in MeOH (44 ml) were treated at room temperature for 1 h according to the similar procedure as described for the preparation of 59 to afford 76 (916.3 mg, quant) as a colorless solid. [α]D22 +75.4° (c 0.96, MeOH); ESI–MS m/z 603 (M+H)+ as C30H42N4O5S2; TOF–ESI–HR-MS (M+H)+ calcd for C30H42N4O5S2: 603.2675, found: 603.2672; IR (KBr) cm−1 1077, 1415, 1508, 1653, 2361, 3020 and 3690; 1H NMR (400 MHz, CD3OD) δ −0.06 to 0.04 (m, 2 H), 0.37–0.46 (m, 2 H), 0.63–0.73 (m, 1 H), 1.12-–1.19 (m, 2 H), 1.25–1.38 (m, 2 H), 1.37 (d, J=6.8 Hz, 3 H), 1.44–1.58 (m, 1 H), 1.75–1.84 (m, 1 H), 1.92–2.02 (m, 1 H), 1.94 (s, 3 H), 2.18 (dt, J=12.1, 2.3 Hz, 1 H), 2.30 (s, 3 H), 2.69 (dd, J=11.6, 2.7 Hz, 1 H), 2.95-3.03 (m, 1 H), 3.59 (dd, J=10.2, 3.1 Hz, 1 H), 3.85 (br dd, J=3.1, 0.7 Hz, 1 H), 3.95 (dq, J=6.8, 2.7 Hz, 1 H), 4.11 (dd, J=10.2, 5.6 Hz, 1 H), 4.43 (br dd, J=9.9, 0.7 Hz, 1 H), 4.64 (dd, J=9.9, 2.7 Hz, 1 H), 5.27 (d, J=5.6 Hz, 1 H), 7.52–7.58 (m, 2 H), 7.65–7.72 (m, 2 H), 9.06 (s, 2 H), 9.11 (s, 1 H); 13C NMR (100 MHz, CD3OD) δ 5.1, 9.3, 13.9, 20.8, 30.8, 32.8, 37.3, 38.5, 42.6, 44.6, 44.8, 53.8, 56.8, 69.5, 70.2, 70.8, 70.9, 72.2, 90.3, 128.6, 131.9, 133.0, 135.2, 138.4, 155.9, 157.9 and 176.3.
Methyl (7S)-6-N-((2′S, 4′R)-4′-(cyclopropylmethyl)piperidine-2′-carbonyl)-7-deoxy-7-(4-(1,2,3-thiadiazol-4-yl)phenylthio)-α-thiolincosaminide (77)
Compound 65 (crude) and 2,2,2-trifluoroacetic acid (0.50 ml) were treated at 0 °C for 30 min according to the similar procedure as described for the preparation of 44 to afford 77 (44.3 mg, 51% in 2 steps from 33) as a colorless solid. [α]D27 +80.4° (c 0.36, MeOH); ESI–MS m/z 595 (M+H)+ as C27H38N4O5S3; TOF–ESI–HR-MS (M+H)+ calcd for C27H38N4O5S3: 595.2083, found: 595.2073; 1H NMR (400 MHz, CD3OD) δ 0.01–0.09 (m, 2 H), 0.43–0.54 (m, 2 H), 0.66–0.78 (m, 1 H), 1.15–1.22 (m, 1 H), 1.25–1.45 (m, 3 H), 1.38 (d, J=6.9 Hz, 3 H), 1.77–1.91 (m, 1 H), 1.94 (s, 3 H), 1.96–2.04 (m, 1 H), 2.32–2.41 (m, 1 H), 3.02 (dt, J=13.1, 3.1 Hz, 1 H), 3.37-3.45 (m, 1 H), 3.59 (dd, J=10.2, 3.2 Hz, 1 H), 3.38 (dd, J=12.6, 3.1 Hz, 1 H), 3.89 (dd, J=3.2, 0.9 Hz, 1 H), 3.94 (dq, J=6.9, 2.5 Hz, 1 H), 4.10 (dd, J=10.2, 5.6 Hz, 1 H), 4.47 (dd, J=9.9, 0.9 Hz, 1 H), 4.66 (dd, J=9.9, 2.5 Hz, 1 H), 5.28 (d, J=5.6 Hz, 1 H), 7.49–7.56 (m, 2 H), 8.01–8.09 (m, 2 H), 9.22 (s, 1 H).
Methyl (7S)-6-N-((2′S, 4′R)-4′-(cyclopropylmethyl)-1′-N-methylpiperidine-2′-carbonyl)-7-deoxy-7-(4-(1,2,3-thiadiazol-4-yl)phenylthio)-α-thiolincosaminide (78)
Compound 77 (29.7 mg, 0.050 mmol), 36% aqueous formaldehyde (12 μl, 0.15 mmol), AcOH (17 μl, 0.30 mmol) and NaBH(OAc)3 (31.6 mg, 0.15 mmol) in MeOH (0.5 ml) were treated at room temperature for 30 min according to the similar procedure as described for the preparation of 59 to afford 78 (27.3 mg, 90%) as a colorless solid. [α]D23 +48.9° (c 0.28, MeOH); ESI–MS m/z 609 (M+H)+ as C28H40N4O5S3; TOF–ESI–HR-MS (M+H)+ calcd for C28H40N4O5S3: 609.2239, found: 609.2231; 1H NMR (400 MHz, CD3OD) δ −0.14 to 0.04 (m, 2 H), 0.30–0.40 (m, 2 H), 0.52–0.65 (m, 1 H), 1.00–1.18 (m, 2 H), 1.25–1.38 (m, 2 H), 1.30 (d, J=7.0 Hz, 3 H), 1.50–1.67 (m, 1 H), 1.78–1.88 (m, 1 H), 1.86 (s, 3 H), 1.97–2.06 (m, 1 H), 2.49 (s, 3 H), 2.50–2.64 (m, 1 H), 3.10–3.19 (m, 2 H), 3.51 (dd, J=10.2, 3.2 Hz, 1 H), 3.77 (br dd, J=3.2, 0.9 Hz, 1 H), 3.84 (dq, J=7.0, 2.6 Hz, 1 H), 4.01 (dd, J=10.2, 5.6 Hz, 1 H), 4.40 (br dd, J=9.9, 0.9 Hz, 1 H), 4.57 (dd, J=9.9, 2.6 Hz, 1 H), 5.18 (d, J=5.6 Hz, 1 H), 7.42–7.49 (m, 2 H), 7.93-7.99 (m, 2 H), 9.14 (s, 1 H).
In vitro antibacterial activity
MIC (μg/ml) was determined by the agar dilution method, which was described in Clinical and Laboratory Standards Institute (M07-07 in 2006). Test strains of S. pneumoniae and S. pyogenes were subjected to seed culture using brain heart infusion agar (Becton Dickinson and Company, Tokyo, Japan) and 5% defibrinated horse blood. Test strains of H. influenzae were subjected to seed culture using sensitivity disk agar-N ‘Nissui’ (Nissui, Tokyo, Japan), 5% defibrinated horse blood, 5 μg ml−1 Hemin and 15 μg ml−1 nicotinamide adenine dinucleotide. A 5 μl portion of cell suspension of the test strains having about 106 CFU ml−1 was inoculated into sensitivity disk agar supplemented with 5% defibrinated horse blood, 5 μg ml−1 Hemin and 15 μg ml−1 nicotinamide adenine dinucleotide, and incubated at 37 °C for 18–22 h. Then, the MIC was measured.
In vitro antibacterial activity (sensitivity distribution against S. pneumoniae of 60 strains)
The MIC for S. pneumoniae was determined by the twofold microdilution broth method using cation-adjusted Mueller–Hinton broth (Difco Laboratories, Detroit, MI, USA) supplemented with 2% lysed horse blood recommended by the Clinical and Laboratory Standards Institute.61 The inoculum was prepared by making a direct colony Suspension, equivalent to a 0.5 McFarland standard, with isolated colonies selected from Mueller–Hinton agar (Difco Laboratories) supplemented with 5% defibrinated sheep blood. Fifty microliters of the adjusted inoculum was added to each well already containing 50 μl of antimicrobial agent in the dilution series. The final test concentration of bacteria was approximately 5 × 104 CFU per well. The MIC was determined as the lowest concentration that prevented visible growth of bacteria after incubation at 35 °C for 20 h.
Neutropenic rat lung infection model
The study and its protocol were complied with Guidelines on the Management of Animal Experiments established by the Pharmaceutical Research Center, Meiji Seika Pharma Co., Ltd and approved by the Animal Experiment Management Committee of it. Rats used in this study are 6-week-old, specific-pathogen-free, male Sprague-Dawley rats (SD-rats) (n=3) (Charles River Laboratories Japan, Inc., Kanagawa, Japan) weighing 160–180 g. These rats were bred under controlled conditions (temperature, 21–25 °C; humidity, 50–70%; lighting hours, 0700–1900 h), and feed (CRF-1; Oriental Yeast Co., Ltd, Tokyo, Japan) and water were available ad libitum. The rats were allowed to acclimatize for 1 week before the study. The rats were rendered neutropenic by intraperitoneal administration of cyclophosphamide (Sigma-Aldrich) 4 days and the day before infection (80 mg kg−1 of body weight). The rats were infected with S. pneumoniae by the injection of 106 CFU into lung through trachea under anesthesia with mixture of ketamine hydrochloride and xylazine hydrochloride (5:1) by injection intramuscularly. The rats were treated by subcutaneous administration of the test compound at 2 h after infection and were killed 24 h after infection by injection of excessive amounts of pentobarbital. The lung was removed and homogenized. Each homogenate was diluted 10-fold serially with physiological saline and an aliquot of each initial homogenate and dilution series was smeared onto a plate of brain heart infusion agar with 5% horse blood. After was incubation at 35 °C for 24 h, the number of colonies grown on the plate was counted. The detection limit was set at <2.0 log10 CFU per lung; if no colonies were detected in the initial homogenate, the value of 2.0 log10 CFU per lung was adopted. The data were expressed as the mean±s.d. log10 CFU per lung.
Dedication
Dedicated to Professor K. C. Nicolaou and his outstanding contributions to complex natural product total synthesis and chemical biology.

Synthesis of substituted piperidines and 2,3,6,7-tetrahydro-1H-azepine. Conditions: (a) bromocyclopropane, lithium diisopropylamide, THF, −78 °C, 1 h; (b) m-chloroperoxybenzoic acid (mCPBA), CH2Cl2, 0 °C to r.t., 1 h; (c) TMSCN, Me2NCOCl, CH2Cl2, 20 °C 40 min, then r.t.,17 h; (d) 5 N NaOH, MeOH, 50 °C, 8 h; (e) H2, PtO2, AcOH, r.t., 24 h; (f) Boc2O, 2 N NaOH, dioxane, r.t., 15 h; (g) BnBr, iPr2NEt, CH3CN, r.t., 48 h; (h) H2, Pd/C, MeOH, r.t., 1 h; (i) LiOH·H2O, dioxane:H2O=4:1, r.t. 5 h.

Synthesis of key intermediates 30 to 33 and 39. Conditions: (a) N,N′-dicyclohexylcarbodiimide (DCC) or EDC·HCl, HOBt, DMF, r.t., 6–20 h; (b) TMSCl, HMDS, pyridine, r.t., 20 min–1 h; (c) 6 N AcOH or 2 N AcOH, MeOH, r.t., 40 min–6 h; (d) MsCl, NEt3, CH2Cl2, 0 °C to r.t., 1 h; (e) AcSK, DMF, 80 °C, 1.5–3 h; (f) 1 N HCl, MeOH, r.t. or 0 °C, 5–100 min; (g) NaOMe, MeOH, r.t., 15 min–3 h; (h) 4-bromobenzenethiol, Cs2CO3, DMF, r.t., 2 h; (i) H2 (0.95 MPa), Pd/C, MeOH, 40 °C, 70 h; (j) Boc2O, LiOH·H2O, dioxane:H2O=1:1, r.t. 3 h.

Synthesis of novel (7S)-4-(pyrimidin-5-yl)phenylthio LCM derivatives possessing piperidine or azepane as the C-6 side chain. Conditions: (a) 5-(4-bromophenyl)pyrimidine, Pd2(dba)3, Xantphos, iPr2NEt, dioxane, reflux, 2-6 h; (b) TFA, CH2Cl2, −20 °C to r.t., 1.5–6 h.

Synthesis of divergent intermediate 54 and novel LCM derivatives possessing a 4-(2-(dimethylamino)ethyl)phenylthio group at the C-7 position. Conditions: (a) TMSCl, HMDS, pyridine, r.t., 1 h; (b) 6 N AcOH, MeOH, r.t., 15 min; (c) MsCl, NEt3, CHCl3, r.t., 1 h; (d) AcSK, DMF, 80 °C, 1.5 h; (e) TMSCl, HMDS, pyridine, r.t., 3 h; (f) 1 N HCl, MeOH, r.t., 10 min; (g) NaOMe, MeOH, r.t., 15 min; (h) 2-(4-bromophenyl)-N, N-dimethylethanamine, Pd2(dba)3, Xantphos, iPr2NEt, dioxane, reflux, 17 h; (i) 20% aq. KOH, N-benzyl-N, N, N-triethylammonium bromide, r.t., 4 h; (j) 15, EDC·HCl, HOBt, DMF, r.t., 5.5 h; (k) TFA, CH2Cl2, 0 °C, 3.5 h; (l) 36% HCHO, NaBH(OAc)3, AcOH, MeOH, r.t., 1 h.

Synthesis of novel 4′-cis-(cyclopropylmethyl)piperidine LCM derivatives possessing an aliphatic- or aromatic-phenylthio group at the C-7 position. Conditions: (a) the corresponding 4-substituted phenylbromides Pd2(dba)3, Xantphos, iPr2NEt, dioxane, reflux, 4–5 h; (b) 4-methylbenzenesulfonohydrazide, toluene, reflux, 5.5 h; (c) TFA, CH2Cl2, −20 °C to r.t., 0.5–5 h; (d) 36% HCHO, NaBH(OAc)3, AcOH, MeOH, r.t., 0.5–2 h.
References
Reinert, R. R ., van der Linden, M. & Al-Lahham, A. Molecular characterization of the first telithromycin-resistant Streptococcus pneumoniae isolate in Germany. Antimicrob. Agents Chemother. 49, 3520–3522 (2005).
Kim, S. H. et al. Changing trends in antimicrobial resistance and serotypes of Streptococcus pneumoniae isolates in Asian countries: an Asian Network for Surveillance of Resistant Pathogens (ANSORP) study. Antimicrob. Agents Chemother. 56, 1418–1426 (2012).
Ajito, K ., Miura, T ., Furuuchi, T. & Tamura, A. Sixteen-membered macrolides: chemical modifications and future applications. Heterocycles 89, 281–352 (2014).
Morimoto, S ., Takahashi, Y ., Watanabe, Y. & Omura, S. Chemical modification of erythromycins I. Synthesis and antibacterial activity of 6-O-methylerythromycins A. J. Antibiot. 37, 187–189 (1984).
Slobodan, D. et al. Erythromycin series. Part 13. Synthesis and structure elucidation of 10-dihydro-10-deoxo-11-methyl-11-azaerythromycin A. J. Chem. Res. Synop. 40, 152–153 (1988).
Retsema, J. et al. Spectrum and mode of action of azithromycin (CP-62,993), a new 15-membered-ring macrolide with improved potency against Gram-Negative Organisms. Antimicrob. Agents Chemother. 31, 1939–1947 (1987).
Denis, A. et al. Synthesis and antibacterial activity of HMR 3647 a new ketolide highly potent against erythromycin-resistant and susceptible pathogens. Bioorg. Med. Chem. Lett. 9, 3075–3080 (1999).
Clay, K. D. et al. Severe hepatotoxicity of telithromycin: three case reports and literature review. Ann. Intern. Med. 144, 415–420 (2006).
Ross, D. B. The FDA and the case of Ketek. N. Engl. J. Med. 356, 1601–1604 (2007).
Gleason, P. P ., Walters, C ., Heaton, A. H. & Schafer, J. A. Telithromycin: the perils of hasty adoption and persistence of off-label prescribing. J. Manag. Care Pharm. 13, 20–25 (2007).
Department of Health and Human Services. Telithromycin (marketed as Ketek) information available at http://www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm107824.htm Accessed 26 April 2007.
Miura, T. et al. Novel azalides derived from sixteen-membered macrolides. I. Isolation of the mobile dialdehyde and its one-pot macrocyclization with an amine. J. Antibiot. 60, 407–435 (2007).
Miura, T. et al. Novel azalides derived from 16-membered macrolides. III. Azalides modified at the C-15 and 4” positions: improved antibacterial activities. Bioorg. Med. Chem. 18, 2735–2747 (2010).
Mason, D. J ., Dietz, A. & Deboer, C. Lincomycin, a new antibiotic I. Discovery and biological properties. Antimicrob. Agents Chemother. 554–559 (1962).
Magerlein, B. J. & Lincomycin, X. The chemical synthesis of lincomycin. Tetrahedron Lett. 1, 33–36 (1970).
Howarth, G. B ., Szarek, W. A. & Jones, J. K.N. The synthesis of lincomycin. J. Chem. Soc. (c) 16, 2218–2224 (1970).
Perlman, D. Structure-Activity Relationships Among the Semisynthetic Antibiotics. Academic Press: New York, San Francisco, London, A Subsidiary of Harcourt Brace Jovanovich Publishers, 1977, pp 600–651.
Birkenmeyer, R. D. & Kagan, F. Lincomycin. XI. Synthesis and structure of clindamycin. A potent antibacterial agent. J. Med. Chem. 13, 616–619 (1970).
Shan, P. J ., Vakil, N. & Kabakov, A. Role of intravenous immune globulin in streptococcal toxic shock syndrome and Clostridium difficile infection. Am. J. Health Syst. Pharm. 72, 1013–1019 (2015).
Hoeksema, H. Octoses from antibiotics. The Upjohn Company, Kalamazoo, Mich.,Abstr. Pap. Division of Carbohydrate Chemistry, 149th Meet, American Chemical Society. Am. Chem Soc. Detroit, Mich p 9C (1965).
Magerlein, B. J ., Birkenmeyer, R. D. & Kagan, F. Chemical modification of lincomycin. Antimicrob. Agents Chemother. 727–736 (1966).
Sinkula, A. A ., Morozowich, W ., Lewis, C. & Mackellar, F. A. Synthesis and bioactivity of lincomycin-7-monoesters. J. Pharm. Sci. 58, 1389–1392 (1969).
Magerlein, B. J. & Kagan, F. Lincomycin. IX. 7-Thiol and thioamido analogs of lincomycin. J. Med. Chem. 12, 974–977 (1969).
Lewis, J. G. et al Novel Antimicrobial 7-methyl Lincosamides: Prolamide Analogs. 43rd Interscience Conference on Antimicrobial Agents and Chemotherapy Poster F-1388; Washington, DC, USA.
Bannister, B. Modifications of lincomycin involving the carbohydrate portion. Part III. The 7-O-methyl and 6-de-(1-hydroxyethyl) analogues. J. Chem. Soc. Perkin Trans. I 1676–1682 (1973).
Bannister, B. Modifications of lincomycin involving the carbohydrate portion. Part IV. (7S-7-alkoxy-7-deoxy-analogues. J. Chem. Soc. Perkin Trans. I 1974, 360–369 (1974).
Bannister, B. & Mydlow, P. K. The S-alkylation of sulphides by an activated carbohydrate epimine under acidic catalysis: the formation of α-acetamido-sulphides. Part 5. The introduction of functionality into the sulphide substituent. J. Chem. Res. (S) 1989, 90–91 (1989).
Bannister, B. The S-alkylation of sulphides by an activated carbohydrate epimine under acidic catalysis: The formation of α-acetamido-sulphides. Part 4. Reaction with dithioacetals and monothioacetals. J. Chem. Soc. Perkin. Trans. I 1980, 540–552 (1980).
Bannister, B. 7S-7-deoxy-7-substituted-alkylthio-lincomycin. S-Alkylation of sulphides by an activated epimine under acidic catalysis: formation of α-acetamido-sulphides. Tetrahedron 40, 1633–1660 (1984).
Bannister, B. The Upjohn Company. Derivatives of lincomycin and its analogs and process. US Patent US3915954 A (1973).
Bannister, B. The Upjohn Company. Derivatives of lincomycin and its analogs and process. Canadian Patent CA-971956 A1 (1972).
Sztaricskai, F. et al. Semisynthetic modification of antibiotic lincomycin. J. Antibiot. 49, 941–943 (1996).
Umemura, E. et al Lincomycin derivative and antibacterial agent containing the same as active ingredient. Japanese Patent WO/2007/066805 A1 (14 June 2007).
Wakiyama, Y. et al Lincomycin derivatives and antibacterial agents containing the same as the active ingredient. Japanese Patent WO/2008/146917 A1 (4 December 2008).
Umemura, E. et al Lincosamide derivative, and antibacterial agent comprising the same as active ingredient, WO/2008/146919 A1 (4 December 2008).
Umemura, E. et al. Synthesis of novel lincomycin derivatives and their in vitro antibacterial activities. J. Antibiot. 66, 195–198 (2013).
Wakiyama, Y. et al. Synthesis and structure–activity relationships of novel lincomycin derivatives. Part 1. Newly generated antibacterial activities against Gram-positive bacteria with erm gene by C-7 modification. J. Antibiot. 69, 368–380 (2016).
Wakiyama, Y. et al. Synthesis and structure–activity relationships of novel lincomycin derivatives. Part 2. Synthesis of 7(S)-7-deoxy-7-(4-morpholinocarbonylphenylthio)lincomycin and its 3-dimensional analysis with rRNA. J. Antibiot. 69, 428–439 (2016).
Kumura, K. et al. Synthesis and antibacterial activity of novel lincomycin derivatives. I. Enhancement of antibacterial activities by introduction of substituted azetidines. J. Antibiot. 69, 440–445 (2016).
Wakiyama, Y. et al. Synthesis and structure–activity relationships of novel lincomycin derivatives Part 3: discovery of the 4-(pyrimidin-5-yl)phenyl group in synthesis of 7(S-thiolincomycin analogs. J. Antibiot. 70, 52–64 (2017).
Kumura, K. et al. Synthesis and antibacterial activity of novel lincomycin derivatives. II. Exploring (7S-7-(5-aryl-1,3,4-thiadiazol-2-yl-thio)-7-deoxylincomycin derivatives. J. Antibiot. 70, 655–663 (2017).
Wakiyama, Y. et al. Synthesis and structure-activity relationships of novel lincomycin derivatives Part 4. Synthesis of novel lincomycin analogs modified at the 6- and 7-positions and their potent antibacterial activities. J. Antibiot. 70, 888–906 (2017).
Kumura, K. et al Synthesis and antibacterial activity of novel lincomycin derivatives. III. Optimization of a phenyl thiadiazole moiety. J. Antibiot. (e-pub ahead of print 5 July 2017; doi:10.1038/ja.2017.59.
Argoudelis, A. D ., Coats, J. H ., Mason, D. J. & Sebek, O. K. Microbial transformation of antibiotics. III. Conversion of clindamycin to 1′-demethylclindamycin and clindamycin sulfoxide by Streptomyces species. J. Antibiot. 22, 309–314 (1969).
Magerlein, B. J ., Birkenmeyer, R. D. & Kagan, F. Lincomycin. VI. 4′-alkyl analogs of lincomycin. Relationship between structure and antibacterial activity. J. Med. Chem. 10, 355–359 (1967).
Magerlein, B. J. Lincomycin. 14. An improved synthesis and resolution of the antimalarial agent, 1′-demethyl-4′-depropyl-4′ (R- and -(S-pentylclindamycin hydrochloride (U-24,729A). J. Med. Chem. 15, 1255–1259 (1972).
Magerlein, B. J. & Lincomycin. VII. 4′-depropyl-4′-ethoxylincomycins. J. Med. Chem. 10, 1161–1163 (1967).
Lewis, J. G. et al Novel lincomycin derivatives possessing antimicrobial activity. WO/2006/055070 A2 (26 May 2006).
Birkenmeyer, R. D ., Kroll, S. J ., Lewis, C ., Stern, K. F. & Zurenko, G. E. Synthesis and antimicrobial activity of clindamycin analogues: pirlimycin, a potent antibacterial agent. J. Med. Chem. 27, 216–223 (1984).
O’Dowd, H. et al. Novel antibacterial azetidine lincosamides. Bioorg. Med. Chem. 18, 2645–2648 (2008).
O’Dowd, H. et al Novel antibacterial azetidine lincosamides. 44 the Interscience Conference on Antimicrobial Agents and Chemotherapy. Poster F-2037; Washington, DC, USA, 2004.
Lewis, J. G. et al Novel antimicrobial 7-methyl lincosamides: Pipecolamide analogs. 43rd Interscience Conference on Antimicrobial Agents and Chemotherapy. Poster F-1389; Washington, DC, USA, 2004.
Chen, T. et al Novel 4′-cycloalkyl pipecolamide lincosamide analogs. 44 th Interscience Conference on Antimicrobial Agents and Chemotherapy. Poster F-2036; Washington, DC, USA, 2004.
Lopez, S. L. et al Characterization of the spectrum of in vitro activity of VIC-105555, a new lincosamide. 44 th Interscience Conference on Antimicrobial Agents and Chemotherapy. Poster F-2038; Washington, DC, USA, 2004.
Shuman, R. T ., Ornstein, P. L ., Paschal, J. W . & Gesellchen, P. D . An improved synthesis of homoproline and derivatives. J. Org. Chem. 55, 738–741 (1990).
Schroeder, W ., Bannister, B. & Hoeksema, H. Lincomycin. III. The structure and stereochemistry of the carbohydrate moiety. J. Am. Chem. Soc. 89, 2448–2453 (1967).
Houtman, R. L. & Mich, P. The Upjohn Company. Trimethylsilyl ethers of lincomycin and its compounds. US Patent US3418414 (1966).
Itoh, T. & Mase, T. A general palladium-catalyzed coupling of aryl bromides/triflates and thiols. Org. Lett 6, 4587–4590 (2004).
Magerlein, B. J. & Kagan, F. Lincomycin. 8. 4′-Alkyl-1′-demethyl-4′-depropylclindamycins, potent antibacterial and antimalarial agents. J. Med. Chem. 12, 780 (1969).
Farrell, D. J ., Morrissey, I ., Bakker, S. & Felmingham, D. Molecular characterization of macrolide resistance mechanisms among Streptococcus pneumoniae and Streptococcus pyogenes isolated from the PROTEKT 1999-2000 study. J. Antimicrob. Chemother. 50 (suppl_2), 39–47 (2002).
Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing; Sixteenth Informational Supplement. CLSI Document M100-S16, Clinical and Laboratory Standards Institute, Wayne, PA, (2006).
Acknowledgements
We thank Mr A. Tamura, Dr E. Shitara and Dr T. Yoshida for encouragement and valuable discussion. We are grateful to Professor Emeritus Dr M. Konno for supervision through our in-house drug discovery program in LCM field. We also thank Dr T. Murata for ROESY analysis; Ms M. Ishii for direction in intellectual properties; Mr T. Watanabe for computational chemistry; Ms T. Miyara, Ms S. Miki and Ms K. Kaneda for analytical and synthetic chemistry; Mr Y. Takayama and Ms K. Yamada for biological studies; and Ms M. Takagi and Ms Y. Saito for English manuscript.
Author information
Authors and Affiliations
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article

Cite this article
Wakiyama, Y., Kumura, K., Umemura, E. et al. Synthesis and SARs of novel lincomycin derivatives Part 5: optimization of lincomycin analogs exhibiting potent antibacterial activities by chemical modification at the 6- and 7-positions. J Antibiot 71, 298–317 (2018). https://doi.org/10.1038/ja.2017.114
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2017.114
This article is cited by
-
Characterization of compound A, a novel lincomycin derivative active against methicillin-resistant Staphylococcus aureus
The Journal of Antibiotics (2021)
