
Abstract
Two new benzoxazines were isolated from Streptomyces griseus (HKI 0545) and assigned as chandrananimycin E (1) and dandamycin (2). Although a number of phenoxazinone-type compounds have been reported from nature, phenoxazines are rarer, and carbon substitution at N-10 such as in 1 is unprecedented. The cyclopentene-containing ring structure of dandamycin (2) is also unique. Chandrananimycin E (1) was found to possess moderate antiproliferative activity against HUVEC cells (GI50 35.3 μM) and weak cytotoxic activity towards HeLa cells (CC50 56.9 μM). Dandamycin showed neither antiproliferative activity nor cytotoxicity towards these cell lines. Structure activity comparisons with phenoxazinones isolated from S. griseus HKI 0545 suggested that the alteration of the core ring systems in 1 and 2 diminishes their activity. Natural products 1 and 2 are interesting additions to the rich secondary metabolome of S. griseus and constitute an important addition to the body of knowledge on phenoxazinone-derived metabolites.
Similar content being viewed by others
Introduction
Natural products featuring a phenoxazinone chromophore exhibit various biological activities including anticancer, antibacterial, antiviral and antifungal.1, 2, 3 Although there are examples of phenoxazinones having been isolated from fungi, insects and even marsupials, they are primarily produced by bacteria, in particular actinomycetes.1 The most well-known members of this group are the actinomycin-type chromopeptides. The actinomycins are potent antibiotics, but the side effects associated with their strong ability to intercalate DNA limit their therapeutic use.3, 4 Various non-peptidic phenoxazinones have been shown to share the actinomycin ability to intercalate DNA as well as their antibiotic and cytotoxic properties.1, 4 Streptomyces spp. have been found to produce a large family of phenoxazinone metabolites, including exfoliazone,5 the elloxazinones,6 the grixazones,7 the chandrananimycins2 and the venezuelines.8 Typically, these compounds differ in their substitution pattern (usually alcohol, amino and carbonyl functions) at positions C-1, C-2, C-8 and C-9 of the phenoxazinone core.
Although phenoxazinone-type molecules are widely distributed in nature, those possessing a phenoxazine core are somewhat rarer. Phenoxazines share the tricyclic iminoquinone core of the phenoxazinones, however, they lack the unsaturated ketone that typifies the latter compounds. Examples of naturally occurring phenoxazine derivatives include venezueline C from Streptomyces venezuelae8 and pycnosanguin from Pycnoporus sanguineus.9
Previous work had shown that a Streptomyces griseus strain (HKI 0545) isolated from the plaster of an old, moisture-damaged building was capable of producing a diverse set of aromatic secondary metabolites. These included several antiproliferative phenoxazinones, such as chandrananimycin D (3),10 bezerramycins A–C (4–6),11 pitucamycin (7),10 grixazone B (8)7, 10 and elloxazinone B (9).5, 11 Furthermore, the quinazoline farinamycin was discovered from the same strain upon variation of the fermentation conditions.12 Recent investigations of the strain HKI 0545 led to the isolation of two previously undetected metabolites, which after structure elucidation were found to possess unusual, altered phenoxazinone-type ring systems. These two compounds were assigned as chandrananimycin E (1, 34 mg) and dandamycin (2, 2.5 mg) (Figure 1).

Structures of chandrananimycin E (1), dandamycin (2), chandrananimycin D (3), bezerramycins A–C (4–6), pitucamycin (7), elloxazinone B (8), grixazone B (9), the methylated chandrananimycin E derivative 10 and venezueline D (11). In case of 2 and 7, the relative configuration is depicted.
Results and Discussion
A large-scale fermentation (300 l, soybean/glucose medium) of Streptomyces griseus (HKI 0545) was undertaken for 5 days before the culture supernatant was separated from the mycelium by filtration and subjected to an amberchrom-161M resin column (CH3OH/H2O). The eluate was concentrated under reduced pressure and lyophilized before being extracted with CH3OH (1 l). The crude mixture obtained was separated by column chromatography on silica gel (CHCl3/CH3OH). Analysis by TLC suggested the presence of two potential phenoxazine derivatives, as indicated by a color change to purple when the plates were stained using anisaldehyde spray reagent. The fractions were further purified by repeated column chromatography on silica gel (CHCl3/CH3OH) followed by Sephadex LH-20 (CH3OH) to yield chandrananimycin E (1, 34 mg) and dandamycin (2, 2.5 mg) (Figure 1).
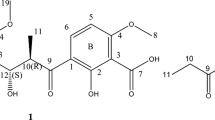
From the HRESIMS data, the molecular composition of C17H16N2O7 was inferred for compound 1, which corresponds to 11 double bond equivalents. The 1H NMR spectrum of 1 contained 12 signals, which in combination with the DEPT and HSQC data could be assigned as four exchangeable protons (δH 9.12, 6.13, 5.29, 5.05), five aromatic resonances (δH 8.32, 7.48, 7.18, 7.14, 6.71) and three methylene moieties (δH 4.47, 4.27, 3.99) (Table 1). The 13C-NMR spectrum of 1 exhibited 17 signals, which in conjunction with the DEPT experiment were found to be five methine (δC 102.9, 115.7, 116.3, 123.0, 125.2), three methylene (δC 60.4, 61.7, 62.3) and nine quaternary carbons (δC 119.1, 121.6, 128.4, 138.2, 145.5, 146.4, 149.2, 170.2, 171.1). The chemical shifts at δC 170.2 and 171.1 ppm pointed to carbonyl moieties. The full structure of 1 was solved by analyses of the 1H-1H COSY, HSQC and HMBC spectra (see Supplementary Figures S1–S6). Examination of the 1H-1H COSY data allowed for the coupling system H-6/H-7 to be established. The HMBC spectrum showed correlations from H-6 (δH 7.14) to C-5a (δC 149.2), C-8 (δC 138.2) and C-9a (δC 128.4), from H-7 (δH 7.18) to C-5a (δC 149.2) and C-9 (δC 123.0), and from H-9 (δH 7.48) to C-5a (δC 149.2) and C-7 (δC 125.2), leading to the construction of an aromatic system with quaternary carbons at C-5a (δC 149.2), C-8 (δC 138.2) and C-9a (δC 128.4) (ring A, Figure 2). The protons at H-7 (δH 7.18) and H-9 (δH 7.48) also showed HMBC correlations to the methylene at C-15 (δH 4.47, δC 62.3), allowing a short chain to be positioned at C-8 (δC 138.2). A 1H-1H COSY correlation from 16-OH (δH 5.29) to H-15 (δH 4.47) capped this chain with a hydroxy group.

1H-1H COSY (bold lines) and selected HMBC correlations (arrows) for chandrananimycin E (1) and dandamycin (2).
Ring C of 1 was constructed by analysis of the HMBC spectrum, which showed correlations from H-1 (δH 8.32) to C-3 (δC 145.5) and C-4a (δC 146.4) and from H-4 (δH 6.71) to C-2 (δC 121.6) and C-10a (δC 119.1). HMBC signals from 11-NH (δH 9.12) to C-1 (δC 115.7), C-2 (δC 121.6), and C-3 (δC 145.5) allowed it to be positioned at C-2. Further HMBC correlations from 11-NH to C-12 (δC 170.2) and C-13 (δC 61.7) allowed for a hydroxyacetyl moiety to be positioned at the nitrogen, as supported by a HMBC correlation between 14-OH (δH 6.13) and C-12 (δC 170.2) and a shared 1H-1H COSY signal between 14-OH (δH 6.13) and H-13 (δH 3.99). The chemical shift of C-3 (δC 145.5) indicated oxygenation at this site.
The methylene at H-18 (δH 4.27) demonstrated a 1H-1H COSY correlation to 19-OH (δH 5.05). The exchangeable proton (19-OH) in turn possessed HMBC correlations to C-18 (δC 60.4) and C-17 (δC 171.1), allowing for a hydroxyacetyl fragment to be constructed. This moiety demonstrated no further correlations in any of the 2D NMR experiments, suggesting it was attached to the molecule via a heteroatom. As such there were two possible positions for this fragment, either at the oxygen positioned at C-3, or attached to the nitrogen at 10-N. Analysis of the NOESY spectrum showed that H-18 (δH 4.27) possessed correlations to both H-9 (δH 7.48) and H-1 (δH 8.32), allowing for the hydroxyacetyl fragment to be placed at 10-N (see Supplementary Figure S7). To unequivocally confirm this positioning, the methylation of compound 1 followed by NMR analysis of the product was undertaken. The HMBC spectrum of the methylated compound 10 (5.2 mg, 90% yield) possessed signals from the methoxy protons (H-20, δH 3.89) to C-3 (δC 147.3) (see Supplementary Figures S17–S22). This established the structure of compound 1, to which we have assigned the trivial name chandrananimycin E.
Ren et al.8 isolated venezuelines A–E from Streptomyces venezuelae in 2013. Venezueline D (11) can be seen to have a similar structure to chandrananimycin E (1), with the second hydroxyacetyl moiety having been positioned at the oxygen at C-3 as opposed to 10-N. Direct comparison of the 1H NMR and 13C NMR spectra for venezueline D (11), provided in the supporting information of the original publication,8 to the NMR spectra of compound 1 showed that the two compounds possess almost identical NMR data. Thus, it seems that the structure of venezueline D (11) can be reassigned as being the same as that of chandrananimycin E (1).
The molecular formula of dandamycin (2) was determined as C13H13NO4 by HRESIMS data analysis (m/z 248.0917 [M+H]+), which corresponds to eight double bond equivalents. Examination of the 13C NMR spectrum revealed nine sp2-hybridized carbon atoms, which were assigned to a ketone (δC 197.2) and four carbon–carbon double bonds. A resonance at δC 162.8 ppm suggested the presence of a vinylogous amide function. These data indicated that 2 must possess three ring structures in order to comply with the calculated degrees of unsaturation. Twelve discrete signals were observed in the 1H NMR spectrum that could be assigned as an amine (δH 10.83), two hydroxyl (δH 5.19, 4.78), three aromatic (δH 6.97, 6.94, 6.85), one olefinic (δH 5.13), two methine protons (δH 4.74, 2.51) and two methylene groups (δH 4.41, 3.71/3.81) according to the HSQC and DEPT experiments (Table 2). Much like in the case of compound 1, analyses of the 1H-1H COSY and HMBC data unveiled an isolated AMX system with coupling constants in the 1H NMR spectrum characteristic of a 1,2,4-trisubstituted benzene moiety as well as a hydroxymethylene residue (see Supplementary Figures S8–S15). These two moieties could be linked because of HMBC correlations from H-7 (δH 6.97) and H-9 (δH 6.85) to C-12 (δC 62.4) (Figure 2). The chemical shifts of the deduced hydroxymethylbenzene residue in combination with HMBC interactions from 6-NH (δH 10.83) to C-6a (δC 127.5) and C-10a (δC 143.9) were suggestive of a 1,4-benzoxazine.
Further analysis of the 1H-1H COSY and HMBC data led to the identification of a 3,4-disubstituted 5-(hydroxymethyl)cyclopent-2-en-1-one moiety. 1H-1H COSY correlations between H-11a/H-3 and H-3/H-2 allowed for this coupling system to be established. HMBC signals from H-11a (δH 4.74) to C-4 (δC 197.2) and C-5 (δC 99.2) positioned the ketone moiety and indicated a cyclopentene ring, as confirmed by further HMBC correlations between H-5 (δH 5.13) and C-3 (δC 53.6), C-4 (δC 197.2) and C-11a (δC 73.4). The positioning of the hydroxymethylene side chain at C-3 (δC 53.6) was confirmed by HMBC interactions between H-2 (δH 3.71/3.81) and C-4 (δC 197.2) and C-11a (δC 73.4). The cyclopentene ring was then connected with the benzoxazine moiety on the basis of a key HMBC interaction from 6-NH (δH 10.83) to C-11a (δC 73.4) to give the planar structure of 2. The relative stereochemistry of the chiral centers at C-3 and C-11a were subsequently deduced from a 2D NOESY experiment (see Supplementary Figure S16). Strong NOE interactions between H-3 (δH 2.51) and H-11a (δH 4.74) suggested both protons reside on the same side of the cyclopentene ring.
The discovery of 1 and 2 in the secondary metabolome of S. griseus is intriguing because these compounds are clearly biogenetically related to the various phenoxazinones previously isolated from the same strain. To evaluate the impact of the irregular architectures of the new compounds on their biological activities, chandrananimycin E (1) and dandamycin (2) were subjected to antiproliferative and cytotoxicity screens (Table 3). Chandrananimycin E (1) was observed to possess moderate antiproliferative activity against the cell line HUVEC (GI50 35.3 μM) and very weak activity towards K-562 (GI50 85.0 μM) cells. Compound 1 also possessed weak cytotoxic activity towards HeLa cells (CC50 56.9 μM). Dandamycin (2) showed neither antiproliferative activities nor cytotoxicity towards the same cell lines.
Our group has previously isolated a number of phenoxazinone derivatives from S. griseus HKI 0545 and tested these natural products for their antiproliferative activities against the cell lines HUVEC and K-562, as well as for their cytotoxicity levels towards HeLa cells. These compounds included chandrananimycin D (3),10 bezerramycins A–C (4–6),11 pitucamycin (7),10 elloxazinone B (8),11 and grixazone B (9)10 (Table 3). When tested against HUVEC cells, chandrananimycin D (3) showed strong antiproliferative activity (GI50 5.3 μM), and bezerramycins A–C (4–6) and pitucamycin (7) showed moderate activity (GI50 14.5–36.6 μM). In activity tests against K-562 cells, chandrananimycin D (3) and pitucamycin (7) showed moderate values (GI50 16.6 and 27.6 μM, respectively), and bezerramycins A–C (4–6) showed weak to no activity (GI50>49.0 μM). Elloxazinone B (8) and grixazone B (9) were inactive against all cell lines tested. All compounds investigated possessed weak to no cytotoxicity towards HeLa cells (CC50 74.8–90.6 μM).
With these activity values in hand, it was possible to propose some structure-activity relationships. When considering activity towards HUVEC cells, it is well conceivable that the intact phenoxazinone architecture is critical for antiproliferative activity. Compounds 1 and 2, with their altered ring structures, both lacked the strong activity of related phenoxazinone derivatives such as chandrananimycin D (3). The N-substitution of 1 may have also reduced its activity. A number of synthetically derived N-substituted phenoxazines have been previously reported and been found to potently inhibit disease pathways associated with, among others,13 neuropathic pain, neurodegenerative diseases,14, 15 type 2 diabetes15, 16 and tumorigenesis,17 and are capable of modulating multidrug-resistant cells in cancer chemotherapy.13, 18
Comparison of bezerramycin A (4) with elloxazinone B (8) would suggest that substitution of the C-15 hydroxymethyl group in the former with a carboxylic acid side chain in the latter has resulted in a direct loss in activity against HUVEC cells. It is likely that the functionality attached at position C-2 also plays a role, as there is a slight activity improvement as the chain length increases when comparing compound 3 through to 6. This may also explain why chandrananimycin E (1) still possesses some activity compared with dandamycin (2), which lacks a similar side chain. The epoxyquinone-type side chain at position C-2 of pitucamycin (7) may be too bulky, resulting in a lowered bioactivity. Substitution at position C-1 may also have a part to play in the bioactivity of these compounds, however, no distinct pattern could be discerned. In regard to activity against K-562 cells, only chandrananimycin D (3, GI50 16.6 μM) and pitucamycin (7, GI50 27.6 μM) possessed activity values of note. All compounds possessed weak to no cytotoxicity towards HeLa cells.
In summary, chandrananimycin E (1) and dandamycin (2) were found to be components of the rich secondary metabolome of S. griseus HKI 0545. Their structure elucidation revealed that the new compounds possess unusual benzoxazine-derived ring systems. When considering compounds from nature, although a number of phenoxazinones have been reported, phenoxazines are rarer, and carbon substitution at 10-N such as in 1 is unprecedented. The unusual ring structure of dandamycin (2) is also unique, there being no similar naturally occurring benzoxazine-type compounds. Chandrananimycin E (1) and dandamycin (2) therefore constitute an important addition to the body of knowledge on phenoxazinone-derived metabolites.
Materials and methods
General experimental procedures
Optical rotation was measured with a JASCO P-1020 polarimeter at 25 °C (JASCO, Gross-Umstadt, Germany). UV spectra were recorded on a Shimadzu UV-1800 spectrophotometer (Shimadzu, Kyoto, Japan) and IR spectra on a JASCO FT-IR (4100) spectrometer. 1H and 13C NMR, DEPT and 2D NMR spectra were measured on Bruker Avance III 500 MHz and Bruker Avance III 600 MHz (equipped with a Bruker Cryo Platform) instruments (Bruker, Fällanden, Switzerland). The chemical shift values (δ) are given in ppm and coupling constants in Hz. 1H and 13C chemical shifts were referenced to the solvent residual peaks for DMSO-d6 at δH 2.49 and δC 39.5, respectively. HRESIMS experiments were carried out on a Thermo Accela UPLC-system combined with a Thermo Exactive mass spectrometer equipped with an electrospray ion source (Thermo Fisher Scientific, Bremen, Germany). Analytical HPLC was performed on an Agilent 1100 Series LC/MSD trap (Agilent Technologies, Santa Clara, CA, USA). Column chromatography was undertaken using silica gel 60 M (230–400 mesh) or Sephadex LH-20 (Pharmacia Biotech AB, Uppsala, Sweden). TLC analyses were performed on silica gel plates (Sil G/UV254 0.20 mm, Macherey-Nagel, Düren, Germany). All solvents used for chromatography, [α]D, UV and MS were HPLC grade.
Bacterial material
Streptomyces griseus (HKI 0545) was isolated from the plaster of an old (built 1953), moisture-damaged building in Herne, Germany. The strain was kindly provided by Dr Wolfgang Lorenz (Institut für Innenraumdiagnostik, Düsseldorf) and taxonomically characterized by the DSMZ (German Collection of Microorganisms and Cell Cultures).
Fermentation and extraction
Liquid organic medium 79 (20 ml, 10 g l−1 dextrose, 10 g l−1 bacto-peptone, 2 g l−1 casein hydrolysate, 2 g l−1 yeast extract, 6 g l−1 NaCl and 15 g l−1 agar) was inoculated with 1 ml suspension of the title strain and incubated at 28 °C on a rotary shaker for 48 h to yield the pre-culture. Subsequently, 3 ml of the pre-culture was transferred to 100 ml of medium 79 (3 × 3 ml of the pre-culture for each 100 ml Erlenmeyer flask). The culture (12 ml) was transferred to 4 l of medium 27 (10 Erlenmeyer flasks with 400 ml of medium 27 each), which was poured into a 300 l-scale fermenter filled with 200 l of medium 27 (20 g l−1 soybean, 20 g l−1 glucose, 5 g l−1 NaCl and 3 g l−1 CaCO3). The fermentation was carried out for 5 days with aeration at 30 l min−1 and stirring at 200 r.p.m. The culture filtrate was separated from the mycelium by filtration and subjected to an amberchrom-161M resin column (20 × 20 cm) using CH3OH/H2O as the eluent (gradient from 10:90 to 90:10 in 35 min). The mycelium and seven fractions of the culture filtrate were dried under reduced pressure and lyophilized.
Extraction and isolation
The culture filtrate (8 g) was extracted with CH3OH (1 l) before the crude extract obtained was separated by column chromatography on silica gel (normal phase) using a gradient from CHCl3 to CH3OH as the eluent. Fractions were further purified by repeated column chromatography on silica gel (CHCl3/CH3OH) and/or Sephadex LH-20 (CH3OH). The compounds of interest were tracked through this process using a combination of analytical HPLC and TLC to yield chandrananimycin E (1, 34 mg) and dandamycin (2, 2.5 mg).
Chandrananimycin E (1): Pale orange solid; UV (CH3OH) λmax (log ɛ) 256 (4.05), 285 (3.88), 304 (3.82) nm; IR (film) 3355, 1666, 1537, 1493, 1436, 1309, 1248, 1078 cm−1; 1H NMR (600 MHz, DMSO-d6): See Table 1; 13C NMR (150.9 MHz, DMSO-d6): See Table 1; HRESIMS m/z 361.1026 [M+H]+ (calcd 361.1030 for C17H17O7N2 [M+H]+).
Dandamycin (2): Yellow gum; [α]D 17.8 (c 0.04, CH3OH); UV (CH3OH) λmax (log ɛ) 235 (4.13), 316 (4.25) nm; IR (film) 3273, 1607, 1578, 1515, 1423, 1386, 1254, 1171, 1047 cm−1; 1H NMR (500 MHz, DMSO-d6): See Table 2; 13C NMR (150.9 MHz, DMSO-d6): See Table 2; HRESIMS m/z 248.0914 [M+H]+ (calcd 248.0917 for C13H14O4N [M+H]+).
Preparation of methylated derivative 10 from chandrananimycin E (1) using TMS-diazomethane
Compound 1 (4.5 mg, 0.0125 mmol) was dissolved in CH3OH:CH2Cl2 (1:1, 1 ml) before TMS-diazomethane (2.0 M in hexanes, 50 μl, 0.1000 mmol) was added drop-wise.19 The reaction was stirred for 1 h at room temperature and then quenched with AcOH (50 μl). The solvent was removed under reduced pressure to give 10 (5.2 mg, 90%).
Compound 10: Brown gum; UV (CH3OH) λmax (log ɛ) 261 (4.23), 285 (4.07), 309 (3.99) nm; IR (film) 3360, 1668, 1537, 1490, 1448, 1428, 1307, 1246, 1080 cm−1; 1H NMR (500 MHz, DMSO-d6) δH 3.89 (s, H-20), 3.99 (s, H-13), 4.29 (s, H-18), 4.48 (s, H-15), 7.02 (s, H-4), 7.15 (d, J=8.3 Hz, H-6), 7.20 (dd, J=1.4, 8.3 Hz, H-7), 7.51 (d, J=1.4 Hz, H-9), 8.39 (s, H-1), 9.10 (s, 11-NH); 13C NMR (150.9 MHz, DMSO-d6) δC 56.5 (C-20), 60.4 (C-18), 61.6 (C-13), 62.2 (C-15), 100.4 (C-4), 115.7 (C-1), 116.2 (C-6), 120.2 (C-10a), 122.1 (C-2), 122.9 (C-9), 125.1 (C-7), 128.2 (C-9a), 138.3 (C-8), 146.6 (C-4a), 147.3 (C-3), 149.1 (C-5a), 170.2 (C-12), 171.1 (C-17); HRESIMS m/z 375.1185 [M+H]+ (calcd 375.1187 for C18H19O7N2 [M+H]+).
Biological assays
Compounds 1 and 2 were assayed for their cytotoxic activities (CC50) against HeLa cells (human cervix carcinoma, DSM ACC 57) and antiproliferative activities (GI50) against the cell lines K-562 (human chronic myeloid cells, DSM ACC 10) and HUVEC (vascular endothelium cells, ATCC CRL-1730) as previously described.20, 21 ICs are provided as 50% inhibition of cell growth (GI50; the concentration needed to reduce the growth of treated cells to 50% of untreated cells) or 50% cytotoxic concentration (CC50; the concentration that kills 50% of treated cells). The initial concentration was 50 μg ml−1.
References
Le Roes-Hill, M., Goodwin, C. & Burton, S. Phenoxazinone synthase: what’s in a name? Trends Biotechnol. 27, 248–258 (2009).
Maskey, R. P., Li, F. C., Qin, S., Fiebig, H. H. & Laatsch, H. Chandrananimycins A-C: Production of novel anticancer antibiotics from a marine Actinomadura sp. isolate M048 by variation of medium composition and growth conditions. J. Antibiot. 56, 622–629 (2003).
Hollstein, U. Actinomycin. Chemistry and mechanism of action. Chem. Rev. 74, 625–652 (1974).
Bolognese, A. et al. Antitumor agents. 1. synthesis, biological evaluation, and molecular modeling of 5H-pyrido[3,2-a]phenoxazin-5-one, a compound with potent antiproliferative activity. J. Med. Chem. 45, 5205–5216 (2002).
Imai, S. et al. Isolation and structure of a new phenoxazine antibiotic, exfoliazone, produced by Streptomyces exfoliatus. J. Antibiot. 43, 1606–1607 (1990).
Graf, E. et al. Elloxazinones A and B, new aminophenoxazinones from Streptomyces griseus Acta 2871. J. Antibiot. 60, 277–284 (2007).
Ohnishi, Y. et al. Structures of grixazone A and B, A-factor-dependent yellow pigments produced under phosphate depletion by Streptomyces griseus. J. Antibiot. 57, 218–223 (2004).
Ren, J. et al. Venezuelines A-G, new phenoxazine-based alkaloids and aminophenols from Streptomyces venezuelae and the regulation of gene target Nur77. Bioorg. Med. Chem. Lett. 23, 301–304 (2013).
Achenbach, H. & Blümm, E. Investigation of the pigments of Pycnoporus sanguineus - pycnosanguin and new phenoxazin-3-ones. Arch. Pharm. 324, 3–6 (1991).
Gomes, P. B., Nett, M., Dahse, H.-M. & Hertweck, C. Pitucamycin: structural merger of a phenoxazinone with an epoxyquinone antibiotic. J. Nat. Prod. 73, 1461–1464 (2010).
Gomes, P. B. et al. Bezerramycins A-C, antiproliferative phenoxazinones from Streptomyces griseus featuring carboxy, carboxamide or nitrile substituents. Eur. J. Org. Chem. 231–235 (2010).
Nett, M. & Hertweck, C. Farinamycin, a quinazoline from Streptomyces griseus. J. Nat. Prod. 74, 2265–2268 (2011).
Thimmaiah, K. N. et al. Synthesis and chemical characterization of N-substituted phenoxazines directed toward reversing vinca alkaloid resistance in multidrug-resistant cancer cells. J. Med. Chem. 35, 3358–3364 (1992).
Hernandez-Olmos, V. et al. N-Substituted phenoxazine and acridone derivatives: Structure−activity relationships of potent P2X4 receptor antagonists. J. Med. Chem. 55, 9576–9588 (2012).
Ebdrup, S. et al. Synthesis and biological and structural characterization of the dual-acting peroxisome proliferator-activated receptor α/γ agonist ragaglitazar. J. Med. Chem. 46, 1306–1317 (2003).
Lohray, B. B. et al. (-)3-[4-[2-(Phenoxazin-10-yl)ethoxy]phenyl]-2-ethoxypropanoic acid [(-)DRF 2725]: A dual PPAR agonist with potent antihyperglycemic and lipid modulating activity. J. Med. Chem. 44, 2675–2678 (2001).
Thimmaiah, K. N. et al. Identification of N10-substituted phenoxazines as potent and specific inhibitors of Akt signaling. J. Biol. Chem. 280, 31924–31935 (2005).
Vock, C. A. et al. Development of ruthenium antitumor drugs that overcome multidrug resistance mechanisms. J. Med. Chem. 50, 2166–2175 (2007).
Barnes, E. C. et al. Antibacterial serrulatane diterpenes from the Australian native plant Eremophila microtheca. Phytochemistry 93, 162–169 (2013).
Huang, X. S. et al. Benzopyrenomycin, a cytotoxic bacterial polyketide metabolite with a benzo[a]pyrene-type carbocyclic ring system. Angew. Chem. Int. Ed. 47, 3995–3998 (2008).
Dahse, H.-M., Schlegel, B. & Grafe, U. Differentiation between inducers of apoptosis and nonspecific cytotoxic drugs by means of cell analyzer and immunoassay. Pharmazie 56, 489–491 (2001).
Acknowledgements
We are grateful to the DBU (Deutsche Bundesstiftung Umwelt) for financial support, Dr Wolfgang Lorenz (Institut für Innenraumdiagnostik, Düsseldorf) for providing the strain, and C. Heiden and M. Steinacker (BioPilot Plant, HKI) for assistance with the fermentations. Dr Hans-Martin Dahse (Department of Infection Biology, HKI) is thanked for undertaking the antiproliferative and cytotoxicity analyses of the isolated compounds. We thank F. Rhein and H. Heinecke for NMR measurements, A. Perner for MS measurements, and U. Valentin and C. Karkowski for technical support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article

Cite this article
Barnes, E., Bezerra-Gomes, P., Nett, M. et al. Dandamycin and chandrananimycin E, benzoxazines from Streptomyces griseus. J Antibiot 68, 463–468 (2015). https://doi.org/10.1038/ja.2015.10
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2015.10
This article is cited by
-
New antimalarial agents derived from nonlinear phenoxazine ring system
Medicinal Chemistry Research (2020)
-
Synthesis, characterization, computational and biological study of novel azabenzo[a]phenothiazine and azabenzo[b]phenoxazine heterocycles as potential antibiotic agent
Medicinal Chemistry Research (2018)
-
Diazabenzo[a]phenoxazone sulphonamides: synthesis, in-silico and in-vitro antimicrobial studies
Medicinal Chemistry Research (2018)
