
Abstract
A new (2) and four known (1, 8–10) sesquiterpenoids, two new (3 and 4) and eight known (5–7, 11–15) xanthone derivatives were isolated from the cultures of sponge-derived fungus Stachybotry sp. HH1 ZDDS1F1-2. The structure of the compounds 1–15 was determined mainly by analysis of the one-dimensional and two-dimensional NMR spectroscopic data and by analogy with the data of those reported. Compound 1 was confirmed by X-ray crystallography. All the compounds were tested for their cytotoxic, antiinflammatory and antiviral (EV71) effects. Compounds 5, 7 and 11 showed significant cytotoxicity against selected human tumor cell lines. Compounds 3, 4 and 11 also displayed significant inhibitory activity against cycloooxygenase (COX-2). Compounds 4, 5 and 11 showed activities against intestinal virus EV71.
Similar content being viewed by others
Introduction
In recent years, marine-derived fungi have attracted considerable attention due to their ability to produce structurally unique and biologically active secondary metabolites.1 We have already reported a great number of chemical constituents from marine sponge.2, 3, 4, 5 Sponge-derived fungi represent a competitive source for new and biologically active natural products. In the past few years, many novel bioactive compounds from the genus Stachybotry displayed antihyperlipidemic,6 plasminogen-modulating activity,7 antiinfluenza virus agents.8 In order to efficiently utilize the marine fungal sources and increase the number of novel bioactive metabolites, we have attempted to explore the chemical diversity of sponge-derived fungi. The crude extract of the fungus Stachybotry sp. HH1 ZSDS1F1-2 was found to exhibit antimicrobial activity. Upon large fermentation of strain Stachybotry sp. HH1 ZSDS1F1-2, compounds 1–15 (Figure 1) were isolated, including a known (1) and a new (2) trichothecene-type sesquiterpenoids, two new (3 and 4) and three known (5–7) grisephenone-type xanthone derivatives, three known eremophilane-type sesquiterpenes (8–10), two known xanthones (11 and 12) and three known griseofulvin derivatives (13–15). Griseophenone, methylxanthone are premise compounds of biosynthesis of griseofulvin.9 Griseofulvin is the first oral antifungal drug and has been in use for many years in medical and veterinary applications,9 but we also found that compounds 5, 7 and 11 showed significant cytotoxicities against selected human tumor cell lines. Compounds 3, 4 and 11 displayed significant inhibitory activity against cycloooxygenase (COX-2), and compounds 4, 5 and 11 showed activity against intestinal virus EV71. Here, we describe the isolation, structure elucidation and biological activities of these compounds.

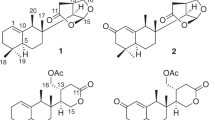
The structures of compounds 1–15.
Results and discussion
Structure elucidation
Compound 1 was isolated as white crystal. The molecular formula of 1 was established as C15H22O4 according to MS and NMR data. The NMR data were in agreement with those previously published 8-hydroxytrichodermol,10 the absolute configuration was confirmed by X-ray crystallography (Figure 2), and this is the first time to report the X-ray data.

The X-ray structure of compound 1
Compound 2 was obtained as pale-white solid. Its positive ion HR–ESI–MS exhibited cationized molecular ion peaks at m/z 265.1422 [M+H]+, 287.1247 [M+Na]+ and 551.2594 [2M+Na]+, corresponding to the molecular formula C15H20O4 (six degrees of unsaturations). The 1H NMR spectrum (Table 1) revealed the presence of three methyl (δH 0.79, 0.84 and 1.85), one tri-substituted C=C bond (δH 5.47, d) and three O-bearing CH groups (δH 4.13, 4.08 and 3.70). The 13C and DEPT–NMR spectra (Table 1) exhibited 16 C-atom signals including those for three methyl (δC 5.3, 16.1 and 18.1), one olefinic (δC 142.4 and 120.5), one C=O group (δC 214.0), three O-bearing CH groups (δC 75.9, 68.5 and 70.8). Apart from one C=O group and one olefinic, there should be four rings in this structure. The general features of the NMR data of 2 (Table 1) resembled those of 1.10 The relative configuration of 2 was determined by NOESY experiment and was assumed to same with 1 by analogy by co-occurring.
Compound 3 was isolated as yellow oil. Its positive ion HR–ESI–MS exhibited cationized molecular ion peaks at m/z 305.1021 [M+H]+, 327.0842 [M+Na]+ and 631.1787 [2M+Na]+, corresponding to the molecular formula C16H16O6 (nine degrees of unsaturations), which was in agreement with the 1H and 13C NMR data (Table 1). The 1H and 13C NMR spectra of 3 were very similar to those of 2,4,4-trihydroxy-2′,4′ -dimethoxy-6′-methylbenzophenone (6).11 The 1H NMR spectrum revealed the presence of one methyl (δH 2.09, s), two methoxyl (δH 3.60, 3.84), one tri-substituted C=C bond (5.47, d) and four aromatic hydrogen groups (δH 6.25, 6.28, 5.09, 5.09). The 13C and DEPT–NMR spectra exhibited 16 C-atom signals including those for one methyl (δC17.7), two methoxyls (δC 54.4, 54.7), one C=O group (δC 200.6), 12 aromatic carbon atoms (δC 135.3, 108.3, 157.3, 96.0, 158.2, 124.6, 106.4, 167.0, 92.7, 164.5, 92.7 and 167.0). Apart from one C=O group, there should be two benzene rings in the structure. HMBC correlations of H-14 with C-2, C-6, C-1; H-2 with C-14, C-4, C-6; H-4 with C-2, C-6, C-5; H-11′ with C-11 and H-3′ with C-3 led to the planar structure of 3.
Compound 4 was isolated as yellow amorphous powder. Its positive ion HR–ESI–MS exhibited cationized molecular ion peaks at m/z 339.0630 [M+H]+, 361.0451 [M+Na]+ and 699.1010 [2M+Na]+, corresponding to the molecular formula C16H15ClO6 (nine degrees of unsaturations), which was in agreement with the 1H and 13C NMR data (Table 1). The 1H and 13C NMR spectra of 4 were very similar to those of grisephenone A (5),12 with the exception of the additional methoxyl moiety. The 1H NMR spectrum character is very same as compound 3, but only three aromatic hydrogens in the low field. HR–ESI–MS also showed there was a chlorine atom in the structure.
Compounds 1–15 were evaluated for their cytotoxicity against 10 human tumor cell lines. Compound 5 showed cytotoxicities against U937 and Hela cell lines, with 50% inhibitory concentration (IC50) values of 22.5 and 14.6 μM. Compound 7 exhibited cytotoxicities against K562 and Hela cell lines, with IC50 values of 22.3 and 14.0 μM. Cytotoxicities for compound 11 against Hela cell line were observed, with IC50 value of 7.2 μM (Supplementary Table S1). Compounds 3, 4 and 11 also displayed significant inhibitory activity against cycloooxygenase (COX-2) with IC50 values of 10.6, 8.9 and 34.3 μM, and compounds 4, 5 and 11 showed activities against intestinal virus EV71 with IC50 values of 30.1, 50.0 and 40.3 μM.
Experimental procedure
General
Column chromatography (CC): commercial silica gel (SiO2: 100–200 and 200–300 mesh; Qingdao Haiyang Chemical Factory, QingDao, China), Sephadex LH-20 (Amersham Biosciences, Uppsala, Sweden) and ODS. Prep.: TLC: glass pre-coated with GF254 SiO2. Medium pressure liquid chromatography: Buchi Sepacore system (Buchi, Flawil, Switzerland). HPLC: Agilent 1260LC system (Santa Clara, CA, USA) with an YMC ODS column (Kyoto, Japan) (10 × 250 mm, 5 μm). One-dimensional and two-dimensional NMR spectra: Bruker AC 500 NMR spectrometer (Brenen, Germany) with TMS as an internal standard. HR–ESI–MS: Bruker micro TOF-QII mass spectrometer. Optical rotation: Anton Paar MCP500 polarimeter (Graz, Austria). IR spectra: Nicolet 6700 FT–IR spectrometer (Waltham, MA, USA). UV spectra: Shimadzu UV-2600 UV–vis spectrophotometer (Tokyo, Japan).
Fungal strain
The fungus (strain no. ZSDS1F1-2) was identified using a molecular biological protocol by DNA amplification and sequencing of the 18 s rRNA gene. By comparison with the GenBank database, the most similar strain was Stachybotry sp. HH1 that are 99% similarity. So the fungus strain ZSDS1F1-2 clearly belongs to the genus Stachybotry, and was designated as Stachybotry sp. HH1.
The fungus strain ZSDS1F1-2 was isolated from fresh healthy sponge which was collected at Xisha Island, China, in April 2012. Following the surface sterilization of the sponge with 75% EtOH, small tissue samples from inside of the sponge were cut aseptically and pressed onto Malt extract agar (malt extract 15 g, sea salt 10 g, chloromycetin 0.2 g, agar 15 g, distilled water 1 l, pH 7.4–7.8) plates. From the growing cultures, pure strain of Stachybotry sp. HH1 was isolated by repeated and reinoculation on MB agar (malt extract 15 g, sea salt 10 g, agar 15 g, distilled water 1 l, pH 7.4–7.8) plates.
Extraction and isolation
Rice solid medium with 3% NaCl (200 g of rice, 200 g water, 6 g sea salt, 20 × 1 l flasks) was inoculated with liquid seed and incubated at 25 °C under daylight for 2 months. The rice matrix and mycelium of Stachybotry sp. HH1 were soaked with acetone for 1 day then broken up with a spatula and extracted three times with EtOAc (3 × 4 l). The combined acetone and EtOAc extract were filtered and evaporated to afford 65.1 g of crude extract.
The crude extract was subjected to silica gel column chromatography (Medium pressure liquid chromatography) eluted with petroleum ether/CHCl3 in gradient eluant (50:50, 0:100) followed by CHCl3/MeOH in gradient eluant (99:1, 95:5, 90:10, 80:20, 0:100) to obtain 15 fractions (Fs 1–10) on the basis of TLC checking. Fraction 3 was purified by Sephadex LH-20 (CHCl3/MeOH, 1:1) first to give three subfractions (Fs 3.1–3.3). F 3.1 (120.0 mg) was subjected to CC (SiO2; eluting with gradient CHCl3/CH3COCH3 50:1–20:1) to give compound 5 (5.3 mg). F 3.3 was subjected to CC (SiO2; CHCl3/CH3COCH3 30:1), followed by repeated prep. TLC (CHCl3/MeOH 100:1) to yield compounds 9 (6.1 mg) and 10 (4.2 mg). F 4 was purified by Sephadex LH-20 (CHCl3/MeOH, 1:1) first to give five subfractions (Fs 4.1–4.5). F 4.1 (180.0 mg) was further purified by CC on silica gel (CHCl3/MeOH, gradient elution 100:1∼20:1), followed by repeated prep. TLC (CHCl3/MeOH 50:1) to afford compounds 6 (10.2 mg) and 7 (8.4 mg). F 5 was purified by Sephadex LH-20 (CHCl3/MeOH, 1:1) first to give four subfractions (Fs 5.1–5.4). F 5.2 (100.0 mg) was further purified by CC on silica gel (CHCl3/MeOH, 50:1) to give compound 8 (7.2 mg). F 6 was purified by Sephadex LH-20 (MeOH) first to give three subfractions (Fs 6.1–6.3). F 6.1 (50.0 mg) was further purified by CC on silica gel (CHCl3/MeOH, 30:1) to give compound 2 (5.4 mg). F 6.3 (124.2 mg) was further purified by HPLC (MeOH/H2O, 75%) to give compounds 3 (5.9 mg) and 4 (7.8 mg). F 7 was purified by Sephadex LH-20 (MeOH) first to yield two subfractions (Fs 7.1–7.2). F 7.1 (20.0 mg) was further purified by HPLC (MeOH/H2O, 80%) to afford compound 11 (4.6 mg). F 7.2 (32.0 mg) was further purified by HPLC (MeOH/H2O, 75%) to afford compound 12 (5.7 mg). F 8 was purified by Sephadex LH-20 (MeOH) first to give five subfractions (Fs 8.1–8.5). F 8.3 (180.0 mg) was further purified by CC on silica gel (CHCl3/MeOH, gradient elution 50:1–1:1), followed by repeated prep. TLC (CHCl3/MeOH 20:1) to afford compounds 13 (2.4 mg) and 1 (3.5 mg). F 9 was purified by Sephadex LH-20 (MeOH) first to give four subfractions (Fs 9.1–9.4). F 9.3 (160.0 mg) was further purified by CC on silica gel (CHCl3/MeOH, 25:1), further purified by HPLC (MeOH/H2O, 45%) to yield compounds 14 (9.4 mg) and 15 (8.1 mg).
8-Hydroxytrichodermol (1): white crystal; 1H NMR (500 MHz, CD3Cl) δH: 3.80 (1H, d, 5.5 Hz, H-2), 2.61 (1H, dd, 15.5, 7.5 Hz, H-3), 1.92 (1H, d, 15.5 Hz, H-3), 4.32 (1H, t, 2.5 Hz, H-4), 2.21 (1H, dd, 14.5, 6 Hz, H-7), 1.73 (1H, d, 14.5 Hz, H-7), 4.10 (1H, d, 5.5 Hz, H-8), 5.53 (1H, d, 5.5 Hz, H-10), 3.58 (1H, d, 6 Hz, H-11), 3.09 (1H, d, 4 Hz, H-13), 2.83 (1H, d, 3.5 Hz, H-13), 0.83 (3H, s, H-14), 1.04 (3H, s, H-15), 1.86 (3H, s, H-16); 13C NMR (125 MHz, CD3Cl) δC: 78.8 (C-2), 40.2 (C-3), 73.6 (C-4), 49.2 (C-5), 39.3 (C-6), 33.7 (C-7), 67.9 (C-8), 139.5 (C-9), 121.6 (C-10), 70.0 (C-11), 65.8 (C-12), 47.7 (C-13), 6.3 (C-14), 18.9 (C-15), 20.6 (C-16). ESI–MS m/z 267 [M+H]+.
Stachybotrichodermone A (2): pale-white solid;  +1.3 (c 0.1, MeOH); UV (MeOH) λmax (log ɛ) 226 (4.63), 300 (3.71), 328 (3.70); IR νmax 3487, 2924, 1739, 1172, 1045, 999, 945, 833; 1H and 13C NMR data see Table 1; selected HMBC data, H-2→C-12, C-11, C-4; H-3→C-12, C-2, C-4; H-7→C-15, C-6, C-8; H-10→C-16, C-6, C-8; H-11→C-15, C-7, C-10, C-9; H-13→C-12, C-2, C-5; H-14→C-6, C-5, C-12, C-4; H-14→C-6, C-5, C-12, C-4; H-15→C-7, C-6, C-5, C-11; H-16→C-8, C-10, C-9 (Supplementary Figures S1–S6); HR–ESI–MS 265.1422, [M+H]+, calcd for 265.1434.
+1.3 (c 0.1, MeOH); UV (MeOH) λmax (log ɛ) 226 (4.63), 300 (3.71), 328 (3.70); IR νmax 3487, 2924, 1739, 1172, 1045, 999, 945, 833; 1H and 13C NMR data see Table 1; selected HMBC data, H-2→C-12, C-11, C-4; H-3→C-12, C-2, C-4; H-7→C-15, C-6, C-8; H-10→C-16, C-6, C-8; H-11→C-15, C-7, C-10, C-9; H-13→C-12, C-2, C-5; H-14→C-6, C-5, C-12, C-4; H-14→C-6, C-5, C-12, C-4; H-15→C-7, C-6, C-5, C-11; H-16→C-8, C-10, C-9 (Supplementary Figures S1–S6); HR–ESI–MS 265.1422, [M+H]+, calcd for 265.1434.
Stachybogrisephenone A (3): brown oil; UV (MeOH) λmax (log ɛ) 295 (4.93), 215 (4.96), 248 (4.11); IR νmax 3248, 2939, 1581, 1427, 1292, 1161, 1022, 825; 1H and 13C NMR data see Table 1; selected HMBC data, H-2→C-14, C-4, C-6; H-4→C-2, C-6, C-3, C-5; H-10, H-12→C-12, C-8, C-9, C-11; H-14→C-2, C-6, C-1; H-15→C-3; H-16→C-11 (Supplementary Figures S7–S10); HR–ESI–MS 305.1021, [M+H]+, calcd for 305.1033.
Stachybogrisephenone B (4): white power; UV (MeOH) λmax (log ɛ) 339 (4.41), 294 (4.94), 215 (4.99), 326 (4.40), 254 (4.26); IR νmax 3402, 2935, 1570, 1431, 1257, 1165, 1083, 925, 837, 806, 767; 1H and 13C NMR data see Table 1; selected HMBC data, H-2→C-14, C-4, C-6, C-3; H-4→C-2, C-6, C-5; H-10→C-12, C-8, C-9; H-14→C-2, C-6, C-1; H-15→C-3; H-16→C-11 (Supplementary Figures S11–S14); HR–ESI–MS 339.0630, [M+H]+, calcd for 339.0630.
Grisephenone A (5): yellow amorphous powder; The 1H NMR (500 MHz, CD3COCD3) δ: 6.32 (1H, d, J=2.0 Hz, H-2), 6.34 (1H, d, J=2.0 Hz, H-4), 6.27 (1H, s, H-10), 2.06 (3H, s, H-14), 3.62 (3H, s, CH3O-C5), 3.98 (3H, s, CH3O-C9), 3.54 (3H, s, CH3O-C11). 13C NMR (125 MHz, CD3COCD3) δ: 136.2 (s, C-1), 109.4 (d, C-2), 159.1 (s, C-3), 96.8 (d, C-4), 158.1 (s, C-5), 125.5 (s, C-6), 201.0 (s, C-7), 101.7 (s, C-8), 162.5 (s, C-9), 88.7 (d, C-10), 163.2 (s, C-11), 108.5 (s, C-12), 162.1 (s, C-13), 19.1 (q, C-14), 55.7 (q, CH3O-C5), 56.7 (q, CH3O-C9), 56.5 (q, CH3O-C11). The spectra data were similar to those in reference;12 ESI–MS m/z 353 [M+H]+.
2,4,4-Trihydroxy-2′,4′-dimethoxy-6′-methylbenzophenone (6): yellow powder; the 1H NMR (500 MHz, CDCl3) δ: 6.35 (1H, s, H-2), 6.35 (1H, s, H-4), 5.94 (1H, s, H-10), 5.93 (1H, s, H-12), 2.16 (3H, s, H-14), 3.71 (3H, s, CH3O-C5), 3.81 (3H, s, CH3O-C3). 13C NMR (125 MHz, CDCl3) δ: 138.1 (s, C-1), 110.3 (d, C-2), 167.6 (s, C-3), 97.8 (d, C-4), 157.8 (s, C-5), 120.3 (s, C-6), 196.6 (s, C-7), 106.0 (s, C-8), 159.0 (s, C-9), 94.6 (s, C-10), 159.0 (s, C-11), 94.6 (d, C-12), 18.8 (q, C-14), 56.0 (q, CH3O-C5), 55.5 (q, CH3O-C3). The spectra data were similar to those in reference.11 ESI–MS m/z 305 [M+H]+.
5,9,11-Trimethoxy-3,13-dihydroxybenzophenone (7): yellow powder; the 1H NMR (500 MHz, CDCl3) δ: 6.20 (1H, d, J=2.0 Hz, H-2), 6.22 (1H, d, J=2.0 Hz, H-4), 5.80 (1H, s, H-10), 2.07 (3H, s, H-14). 13C NMR (125 MHz, CDCl3) δ: 135.6 (s, C-1), 108.6 (d, C-2), 167.7 (s, C-3), 96.3 (d, C-4), 157.0 (s, C-5), 126.0 (s, C-6), 199.8 (s, C-7), 107.5 (s, C-8), 156.3 (s, C-9), 96.3 (d, C-10), 93.5 (s, C-12), 19.0 (s, C-14), 55.7 (q, CH3O-C5), 55.7 (q, CH3O-C9), 55.6 (q, CH3O-C11). The compound was reported in the reference13 but there were no NMR data. This compound was a new natural compound and the NMR data were also firstly reported. ESI–MS m/z 319 [M+H]+.
2,3-Dihydroxy-eremophila-1(10),7(11),8-trien-8,12-olide (8): brown oil; The 1H NMR (500 MHz, CDCl3) δ: 5.63 (1H, s, H-1), 4.34 (1H, H-2), 3.90 (1H, d, J=4.5 Hz, H-3), 1.78 (1H, dd, J=14.0, 7.0 Hz H-4). 2.84 (1H, d, J=16.5 Hz, H-6), 2.17 (1H, d, J=16.5 Hz, H-6), 5.93 (1H, s, H-9), 1.88 (3H, s, H-13), 1.18 (3H, s, H-14), 1.22 (3H, d, 7 Hz, H-15). 13C NMR (125 MHz, CDCl3) δ: 129.8 (d, C-1), 69.0 (d, C-2), 72.1 (d, C-3), 41.1 (d, C-4), 37.8 (s, C-5), 35.8 (t, C-6), 147.2 (s, C-7), 149.0 (s, C-8), 108.3 (d, C-9), 140.6 (s, C-10), 121.8 (s, C-11), 171.4 (s, C-12), 8.4 (q, C-13), 21.4 (q, C-14), 13.0 (q, C-15). The spectra data were similar to those in reference.14 ESI–MS m/z 263 [M+H]+.
2-Oxo-3-hydroxy-eremophila-1(10),7(11),8-trien-8,12-olide (9): brown oil; the 1H NMR (500 MHz, CD3OD) δ: 6.09 (1H, s, H-1), 3.86 (1H, d, J=3.5 Hz, H-3), 2.20 (1H, m), 3.13 (1H, d, J=17.0 Hz, H-6), 2.49 (1H, d, J=17.0 Hz, H-6), 6.28 (1H, s, H-9), 1.99 (3H, s, H-13), 1.35 (3H, s, H-14), 1.29 (3H, d, J=7.0 Hz). 13C NMR (125 Hz, CD3OD) δ: 125.1 (d, C-1), 198.3 (s, C-2), 73.7 (d, C-3), 43.0 (d, C-4), 39.0 (s, C-5), 33.9 (t, C-6), 146.1 (s, C-7), 153.4 (s, C-8), 105.9 (d, C-9), 163.6 (s, C-10), 124.6 (s, C-11), 170.1 (s, C-12), 8.1 (q, C-13), 22.2 (q, C-14), 10.8 (q, C-15). The spectra data were similar to those in reference.15 ESI–MS m/z 261 [M+H]+.
2-Oxo-3-hydroxy-eremophila-1(10),3,7(11),8-tetraen-8,12-olide (10): brown oil; the 1H NMR (500 MHz, CDCl3) δ: 6.41 (1H, s, H-1), 3.10 (1H, d, J=15.5 Hz, H-6), 2.48 (1H, d, J=15.5 Hz, H-6), 6.25 (1H, s, H-9), 2.02 (3H, s, H-13), 1.35 (3H, s, H-14), 2.06 (3H, d, J=7.0 Hz, H-15). 13C NMR (125 MHz, CDCl3) δ: 124.8 (d, C-1), 179.0 (s, C-2), 144.8 (s, C-3), 130.8 (s, C-4), 42.4 (s, C-5), 31.6 (t, C-6), 144.6 (s, C-7), 152.5 (s, C-8), 106.3 (d, C-9), 160.4 (s, C-10), 125.7 (s, C-11), 169.7 (s, C-12), 9.0 (q, C-13), 29.0 (q, C-14), 11.0 (q, C-15). The spectra data were similar to those in reference.16 ESI–MS m/z 259 [M+H]+.
3,6,8-Trihydroxy-1-methylxanthone (11): yellow powder; the 1H NMR (500 MHz, CD3OD) δ: 6.61 (1H, s, H-2), 6.61 (1H, s, H-4), 6.21 (1H, d, J=2.0 Hz, H-5), 6.11 (1H, d, J=2.0 Hz, H-7), 2.77 (3H, s, H-10). 13C NMR (125 MHz, CD3OD) δ: 143.3 (s, C-1), 115.6 (d, C-2), 159.3 (s, C-3), 100.1 (d, C-4), 163.4 (s, C-4a), 164.6 (s, C-5a), 92.8 (d, C-5), 157.1 (s, C-6), 97.3 (d, C-7), 163.4 (s, C-8), 102.5 (s, C-8a), 182.0 (s, C-9), 22.1 (q, C-10). The spectra data were similar to those in reference.17 ESI–MS m/z 259 [M+H]+.
3,6,8-Trihydroxy-5-chlorin-1-methylxanthone (12): yellow powder; the 1H NMR (500 MHz, CD3OD) δ: 6.72 (1H, d, J=2.0 Hz, H-2), 6.68 (1H, d, J=2.0 Hz, H-4), 6.28 (1H, s, H-7), 2.79 (3H, s, H-10). The spectra data were similar to those in reference.18 ESI–MS m/z 293 [M+H]+.
Dechlorodehydrogriseofulvin (13): white powder; the 1H NMR (500 MHz, CDCl3) δ: 6.07 (1H, d, J=2.0 Hz, H-5), 6.28 (1H, d, J=1.5 Hz, H-7), 5.65 (1H, d, J=1.5 Hz, H-3′), 6.17 (1H, t, J=1.5 Hz, H-5′), 3.91 (3H, s, CH3O-C4), 3.91 (3H, s, CH3O-C6), 3.63 (3H, s, CH3O-C2′), 1.78 (3H, d, J=1.5 Hz, CH3-C6′). 13C NMR (125 MHz, CDCl3) δ: 88.2 (s, C-2), 189.3 (s, C-3), 103.8 (s, C-3a), 159.6 (s, C-4), 89.5 (d, C-5), 170.5 (s, C-6), 93.0 (d, C-7), 176.4 (s, C-7a), 168.1 (s, C-2′), 103.8 (s, C-3′), 186.6 (s, C-4′), 129.2 (d, C-5′), 147.4 (s, C-6′), 56.1 (q, CH3O-C4), 56.1 (q, CH3O-C6), 56.2 (q, CH3O-C2′), 16.6 (q, CH3-C6′). The spectra data were similar to those in reference.9 ESI–MS m/z 317 [M+H]+.
Dechlorogriseofulvin (14): white powder; the 1H NMR (500 MHz, CDCl3) δ: 6.04 (1H, H-5), 6.23 (1H, H-7), 5.35 (1H, H-3′), 3.09 (1H, H-5′), 2.42 (1H, H-5′), 2.77 (3H, s, H-6′), 3.91 (3H, s, CH3O-C4), 3.91 (3H, s, CH3O-C6), 3.63 (3H, s, CH3O-C2′), 0.96 (3H, d, J=6.5 Hz, CH3-C6′). 13C NMR (125 MHz, CDCl3) δ: 90.0 (s, C-2), 192.5 (s, C-3), 104.5 (s, C-3a), 159.2 (s, C-4), 93.4 (d, C-5), 171.4 (s, C-6), 88.6 (d, C-7), 176.2 (s, C-7a), 170.5 (s, C-2′), 104.8 (d, C-3′), 197.3 (s, C-4′), 40.2 (t, C-5′), 36.7 (q, C-6′), 56.2 (q, CH3O-C4), 56.1 (q, CH3O-C6), 56.6 (q, CH3O-C2′), 14.3 (q, CH3-C6′). The spectra data were similar to those in reference.19 ESI–MS m/z 319 [M+H]+.
epi-Griseofulvin (15): yellow powder; the 1H NMR (500 MHz, CDCl3) δ: 6.45 (1H, s, H-5), 5.56 (1H, s, H-3′), 3.03 (1H, dd, J=16.5, 13.5 Hz, H-5′), 2.45 (1H, dd, J=16.5, 4.9 Hz, H-5′), 2.78 (1H, t, J=11.5, 5.0 Hz, H-6′), 4.15 (3H, s, CH3O-C4), 4.02 (3H, s, CH3O-C6), 3.64 (3H, s, CH3O-C2′), 0.95 (3H, d, 6.5 Hz, CH3-C6′). 13C NMR (125 MHz, CDCl3) δ: 89.9 (s, C-2), 192.1 (s, C-3), 104.6 (s, C-3a), 156.5 (s, C-4), 90.6 (d, C-5), 164.4 (s, C-6), 109.2 (s, C-7), 170.8 (s, C-2′), 105.0 (d, C-3′), 196.8 (s, C-4′), 40.0 (t, C-5′), 36.5 (q, C-6′), 57.6 (q, CH3O-C4), 57.0 (q, CH3O-C6), 56.6 (q, CH3O-C2′), 14.2 (q, CH3-C6′). The spectra data were similar to those in reference.19 ESI–MS m/z 353 [M+H]+.
X-ray crystallographic analysis of 1
White crystal of 1 was obtained in MeOH. X-ray diffraction intensity data were collected on a CrysAlis PRO charge-coupled device area detector diffractometer with graphite monochromated Cu Kα radiation (λ=1.54184 Å). The structures were solved by direct methods (SHELXL-97) and refined using full-matrix least squares difference Fourier techniques. All non-hydrogen atoms were refined anisotropically, and all hydrogen atoms were placed in idealized positions and refined as riding atoms with the relative isotropic parameters.
Crystal data of 1
White crystal of C15H22O4, orthorhombic, space group P2(1)2(1)2(1) with a=6.7903(4) Å, b=15.0301(8) Å, c=17.7246(11) Å, V=1808.96(17) Å3, Z=34, T=N/A, μ (Cu Kα)=4.730 mm−1, and F(000)=814. Dcalc=1.4162 g mm−3, 5014 reflections measured (7.72⩽2Θ⩽125.44), 2634 unique (Rint=0.0448, Rsigma=0.0563) which were used in all calculations. The final indices were R1=0.0832, wR2=0.2408 (I>2σ(I)).
Cytotoxic assay
Cytotoxicity was assayed with the CCK8 (Dojindo, Japan) method.20 Cell lines, K562, A549, Huh-7, H1975, MCF-7, U937, BGC823, HL60, Hela and MOLT-4 were purchased from Shanghai Cell Bank, Chinese Academy of Sciences, China. Cells were routinely grown and maintained in mediums RPMI or Dulbecco’s modified eagles medium (DMEM) with 10% fetal bovine serum (FBS) and with 1% penicillin/streptomycin. All cell lines were incubated in a Thermo/Forma Scientific CO2 Water-jacketed incubator (Waltham, MA, USA) with 5% CO2 in air at 37 °C. Cell viability assay was determined by the CCK8 (Dojindo, Kumamoto, Japan) assay. Cells were seeded at a density of 400–800 cells per well in 384-well plates and treated with various concentration of compounds or solvent control. After 72-h incubation, CCk8 reagent was added, and absorbance was measured at 450 nm using Envision 2104 multi-label Reader (Perkin Elmer, Foster City, CA, USA). Dose-response curves were plotted to determine the IC50 values using Prism 5.0 (GraphPad Software, San Diego, CA, USA).
Antiinflammatory assay
The COX-2 enzyme is a well-established target for test compound antiinflammatory evaluation.21 Hence, the compounds isolated were tested for COX-2 inhibitory activity using the COX (ovine) inhibitor screening kit, as per the manufacturer’s instructions. The COX-2 enzyme catalyze the biosynthesis of PGH2 from arachidonic acid. In this assay, the PGF2α produced by the reduction of PGH2 with stannous chloride (SnCl2) was measured in this enzyme immunoassay. The stock solutions were prepared in dimethyl sulfoxide, and the final concentration was set as 10 μM for the isolated compounds. The percentage inhibition was calculated by comparison with control incubations. The IC50 values of the most active compounds were calculated from concentration−inhibition response curves. Celecoxib (Sigma, St Louis, MO, USA) was used as the positive control with an IC50 value of 8.09 nM.
Antiviral (EV71) assay
Antienterovirus 71 (EV71) was assayed on Vero cells with the CCK8 (Dojindo, Japan) method.22 Vero cells (2 × 103 cells per well) were seeded with DMEM medium (2% FBS) into a 384-well plate. After 24 h, 1000-fold serial dilution of the compounds were added in triplicate to the 348-well plate. After incubation at 37 °C for 30 min, a twofold dilution 100 × the 50% tissue culture infectious dose (TCID50) of EV71 virus in DMEM supplemented with 2% FBS was added to each well. The plate was incubated at 37 °C for 72–96 h when the viral control cells showed complete cytopathic effect, the cell survival was quantified using CCK8. The A450 of the well was measured with a microtiter platereader (Envision, PerkinElmer). The IC50 of the testing compound was calculated using the GraphPad Prism software. Ribavirin was used as the positive control with an IC50 value of 0.60 μM.
References
Rateb, M. E. & Ebel, R. Secondary metabolites of fungi from marine habitats. Nat. Prod. Rep. 28, 290–344 (2011).
Huang, R., Peng, Y., Zhou, X., Yang, X. & Liu, Y. A new taurine derivative from South China Sea marine sponge Axinella sp. Nat. Prod. Res. 27, 1537–1541 (2013).
Yang, B., Tao, H., Zhou, X., Lin, X.-P. & Liu, Y. Two new alkaloids from marine sponge Callyspongia sp. Nat. Prod. Res. 27, 433–437 (2013).
Liu, Y. et al. New cyclitol derivative from a sponge Sarcotragus species. Nat. Prod. Res. 25, 648–652 (2011).
Zhou, X. et al. Chemical and biological aspects of marine sponges of the genus Xestospongia. Chem. Biodivers. 7, 2201–2227 (2010).
Li, Y. et al. Chartarlactams A–P phenylspirodrimanes from the sponge-associated fungus Stachybotrys chartarum with antihyperlipidemic activities. J. Nat. Prod. 77, 138–147 (2014).
Koide, H., Hasegawa, K., Nishimura, N., Narasaki, R. & Hasumi, K. A new series of the SMTP plasminogen modulators with a phenylamine-based side chain. J. Antibiot. (Tokyo) 65, 361–367 (2012).
Minagawa, K. et al. Novel stachyflin derivatives from Stachybotrys sp. RF-7260. Fermentation, isolation, structure elucidation and biological activities. J. Antibiot. (Tokyo) 55, 239–248 (2002).
Cacho, R. A., Chooi, Y.-H., Zhou, H. & Tang, Y. Complexity generation in fungal polyketide biosynthesis: a spirocycle-forming P450 in the concise pathway to the antifungal drug griseofulvin. ACS Chem. Biol. 8, 2322–2330 (2013).
Lauren, D. R. et al. Trichothecenes produced by Fusarium crookwellense DAOM 193611. J. Agric. Food Chem. 35, 884–889 (1987).
Hay, J. & Harris, T. Biogenetic-type synthesis of heptaketide natural products: alternariol and lichexanthone. J. Chem. Soc. Chem. Commun. 16, 953b–955b (1972).
Liu, S.-Z. & Zhao, W.-M. Chemical constituents of medicinal fungus Shiraia bambusicola. Chin. Trad. Herb. Drugs 8, 006 (2010).
Arison, B. et al. The delineation of griseofulvin and related systems by nuclear magnetic resonance spectroscopy. J. Am. Chem. Soc. 85, 627–631 (1963).
Tabata, Y. et al. PF1092A, B and C, new nonsteroidal progesterone receptor ligands produced by Penicillium oblatum. II. Physico-chemical properties and structure elucidation. J. Antibiot. 50, 309–313 (1997).
Wu, G., Lin, A., Gu, Q., Zhu, T. & Li, D. Four new chloro-eremophilane sesquiterpenes from an Antarctic deep-sea derived fungus, penicillium sp. PR19N-1. Mar. Drugs 11, 1399–1408 (2013).
Kurihara, K. Synthesis of (±)-PF1092A, B and C; new nonsteroidal progesterone receptor ligands. J. Antibiot. 50, 360–362 (1997).
Yang, J., Qiu, S., She, Z. & Lin, Y. A new xanthone derivative from the marine fungus Phomopsis sp. (No. SK7RN3G1). Chem. Nat. Compd. 49, 246–248 (2013).
Elix, J. A., Musidlak, H. W., Sala, T. & Sargent, M. V. Structure and synthesis of some lichen xanthones. Aust. J. Chem. 31, 145–155 (1978).
Shang, Z., Li, X.-M., Li, C.-S. & Wang, B.-G. Diverse secondary metabolites produced by marine-derived fungus nigrospora sp. ma75 on various culture media. Chem. Biodivers. 9, 1338–1348 (2012).
Wanka, L., Iqbal, K. & Schreiner, P. R. The lipophilic bullet hits the targets: medicinal chemistry of adamantane derivatives. Chem. Rev. 113, 3516–3604 (2013).
Bairwa, K. et al. Rotenoids from Boerhaavia diffusa as potential anti-inflammatory agents. J. Nat. Prod. 76, 1393–1398 (2013).
Ho, H-Y, Cheng, M-L, Weng, S-F, Leu, Y-L & Chiu, DT-Y Antiviral effect of epigallocatechin gallate on enterovirus 71. J. Agric. Food Chem. 57, 6140–6147 (2009).
Acknowledgements
This work was supported financially by the National Key Basic Research Program of China (973)’s Project (2010CB833800 and 2011CB915503), the 863 Program (2012AA092104 and 2013AA092902), the National Natural Science Foundation of China (Nos. 52102308, 31270402, 21172230, 20902094, 41176148, 21002110, 31270402, 21172094 and 41376155), Guangdong Province-CAS Joint Research Program (2011B090300023 and 2012B091100264), and Guangdong Marine Economic Development and Innovation of Regional Demonstration Project (GD2012-D01-001 and GD2012-D01-002).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article

Cite this article
Qin, C., Lin, X., Lu, X. et al. Sesquiterpenoids and xanthones derivatives produced by sponge-derived fungus Stachybotry sp. HH1 ZSDS1F1-2. J Antibiot 68, 121–125 (2015). https://doi.org/10.1038/ja.2014.97
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2014.97
This article is cited by
-
Perceiving SARS-CoV-2 Mpro and PLpro dual inhibitors from pool of recognized antiviral compounds of endophytic microbes: an in silico simulation study
Structural Chemistry (2022)
-
Recent progresses in marine microbial-derived antiviral natural products
Archives of Pharmacal Research (2020)
-
Marine natural products with anti-inflammatory activity
Applied Microbiology and Biotechnology (2016)
