
Abstract
Two new cytochalsians, multirostratin A (1) and 20-oxo-deoxaphomin (2), together with five known analogues (3–7), were obtained from the endophytic fungus Phoma multirostrata EA-12. The structures of 1 and 2 were elucidated by MS and 1D- and 2D-NMR spectroscopic data analyses, as well as by comparison of data with those of analogues reported in the literature. Compounds 1 and 2 showed moderate cytotoxicity against five tumor cell lines (HL-60, A-549, SMMC-7721, MCF-7 and SW-480).
Similar content being viewed by others
Introduction
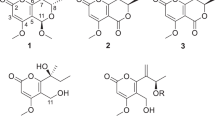
Endophytic fungi have been proved to be a productive source of structurally diverse and biologically active secondary metabolites.1, 2, 3 In our effort to search for structurally interesting and bioactive natural products, >200 strains of endophytic fungi isolated from Eupatorium adenophorum were preliminarily screened for their toxicity against brine shrimp (Artemia salina). In these investigated fungi, one strain, named EA-12, identified as Phoma multirostrata was found lethal to brine shrimp. Chemical investigation on cultures of P. multirostrata led to the isolation of two new cytochalsians, multirostratin A (1) and 20-oxo-deoxaphomin (2), together with five known analogues, deoxaphomin (3),4 cytochalasin A (4),5 cytochalasin B (5),5 cytochalasin Z2 (6)6 and cytochalasin F (7) (Figure 1).7 The new structrures were elucidated by extensive spectroscopic methods, while the known analogues were identified by comparison data with those reported in the literature. Compound 1 was the first cytochalasin with a macrocyclic ring including a furan moiety. The new compounds 1 and 2 showed significant cytotoxicities against five tumor cell lines of HL-60, A-549, SMMC-7721, MCF-7 and SW-480.

Structures of cytochalasins (1–7) from the endophytic fungus Phoma multirostrata.
Results and discussion
Compound 1 was obtained as white, amorphous solid, which was assigned the molecular formula C29H35NO3 from its HR-EI-MS (m/z 445.2622 [M]+, calcd for C29H35NO3, 445.2617), indicating 13° of unsaturation. Its IR spectrum exhibited absorption bands for a hydroxy (3424 cm−1), amide carbonyl (1700 cm−1), olefinic bond (1641 cm−1) and phenyl (1606, 1023, 701 cm−1) groups. 1D NMR and HSQC spectra of 1 revealed 29 carbon signals, including 2 methyls, 6 methylenes (one exo-methylene), 15 methines (one oxymethine) and 6 quarternary carbons (including three carbonyls) (Table 1). The 1H NMR spectrum showed two methyl signals at δH 0.92 (3H, d, J=6.9 Hz, H-24) and 0.96 (3H, d, J=6.7 Hz, H-11), two exocyclic CH2= group at δH 5.10 (1H, s, H-12a) and 5.28 (1H, s, H-12b), a double bond group at δH 5.43 (1H, m, H-14) and 6.07 (1H, ddd, J=15.4, 9.8, 1.5 Hz, H-13) and a monosubstituted phenyl group at δH 7.12 (2H, d, J=7.0 Hz, H-2′, 6′), 7.23 (1H, t, J=7.0 Hz H-4′) and 7.30 (2H, d, J=7.0 Hz, H-3′, 5′). In addition, the presence of a 2,5-disubstituted furan moiety in 1 was assumed by the NMR signals at δH 5.93 (1H, d, J=3.0 Hz, H-21), 6.10 (1H, d, J=3.0 Hz H-22) and at δC 106.2 (C-22), 107.2 (C-21), 153.2 (C-23) and 156.2 (C-20).
Analysis of the 1H–1H COSY spectrum gave two C-linkage fragments: C-10/C-3/C-4/C-5/11-Me and C-7/C-8/C-13/C-14/C-15/C-16/C-17 or 24-Me/C-18/C-19, as shown in bold in Figure 2. In the HMBC spectrum (Figure 2), the correlation of H-4 and H-8 to C-9, C-1 and C-23 indicated that C-1, C-4, C-8 and C-23 were all connected to C-9, and the correlation of H-12 to C-5, C-6 and C-7, together with H-3 to C-3, C-4 and C-5, established an isoindolone moiety in 1. Furthermore, the HMBC correlation of H-21 and H-22 to C-8, C-9, C-19 and C-20, indicating a furan moiety located between C-19 and C-23 (Figure 2). The monosubstituted phenyl group was connected to the isoindolone moiety via C-10 on the correlations of aromatic proton signal at δH 7.12 (H-2′), with the carbon signal at δC 44.2 (C-10). Thus the planar structure of 1 was established.

Selected 2D NMR correlations of 1 and 2.
The relative configuration of 1 was established by a combination of coupling constant and ROESY data. In the 1H NMR spectrum, the large coupling constant (J=10.2 Hz) for H-7 and H-8 suggested the axial–axial orientation of the proton pair. The E-geometry of the Δ13-double bond was deduced from the large coupling constant observed for H-13 (J=14.7 Hz). In the ROESY spectrum, the correlations of H-3/H3-11, H-5/H-4 and H-5/H-8 were observed, which indicated that H-3, H3-11 and H-7 were in the same orientation (α), whereas H-4, H-5 and H-8 were in the opposite direction (β) (Figure 2). The correlations of H-7/H-13, H-14/H-15b, H-15b/H-14, H-15b/H-16 and H-15a/H3-24 indicated that H-13 and H3-24 were α-oriented, while H-14 and H-16 were β-oriented. Consequently, the structure of 1 was established and given the name multirostratin A.
Compound 2 was obtained as white, amorphous solid. Its molecular formula was determined as C29H35NO3 from the positive HREIMS molecular ion peak at m/z 461.2587 [M]+, indicating 13° of unsaturation. The NMR spectral data of 2 (Table 1) resembled to the known analogue deoxaphomin (3) except that the 13C signal at 74.0 ppm (C-20) in 3 was changed to 204.2 p.p.m. in 2, suggesting that a keto carbon atom instead of an oxygen-bearing methine existed in 2. In addition, the 1H signal of H-21 was split into a doublet (J=16.2 Hz) instead of a double doublet (J=15.3, 8.1 Hz) in 3. This was in accordance with the HMBC correlations from H-21 (δH 7.70 (1H, d, J=16.2 Hz)) and H-22 (δH 6.85 (1H, d, J=16.2 Hz)) to C-20 (δC 204.2) and C-19 (δC 41.5) (Figure 2). The other correlations in the HMBC and 1H–1H COSY spectra as shown in Figure 2 also confirmed the connectivities in compound 2. The relative configuration of 2 was established by a combination of coupling constant and ROESY experiment. In the 1H NMR spectrum, the large coupling constant (J=10.5 Hz) for H-7 and H-8 suggested the axial–axial orientation of the proton pair. The E-geometry of the Δ13 and Δ21-double bonds was deduced from the large coupling constant observed for JH-13,H-14=15.2 Hz and JH-21,H-22=16.2 Hz. The ROESY spectrum of 2 displayed similar patterns to those of 1, which suggested that the relative configurations of the chiral centers in 2 were the same to those of 1 (Figure 2). Consequently, compound 2 was identified as 20-oxo-deoxaphomin.
Compounds 1 and 2 showed a certain cytotoxic activities against HL-60, A-549, SMMC-7721, MCF-7 and SW-480, the IC50 values varied between 7.7 and 15.8 μM, as listed in Table 2.
In summary, cytochalasins are a group of polyketide synthases-non ribosomal peptide synthetases (PKS-NRPS) hybrid fungal metabolites that have been isolated from different genera of fungi, such as Phomopsis,8 Aspergillus,9 Spicaria,10 Xylaria,11 Periconia,12 etc. They display a wide range of biological activities, including cytotoxic,11, 12 antibiotic,13 inhibition of HIV-1 protease14 and phytotoxic activities;15 their most unusual property is the ability to cause cells to extrude their nuclei, leading to the formation of nuclei free cells. In general, cytochalasins are structurally characterized by a highly substituted isoindolone fused with an 11- to 13-membered carbocyclic or 12- to 14-membered lactone rings, and most recently, an unprecedented 9-membered carbocyclic rings.12 To date, >100 cytochalasins and derivatives have been isolated from a range of fungi. To the best of our knowledge, multirostratin A (1) was the first example with a macrocyclic ring including a furan moiety.
Methods
General experimental procedures
Optical rotations were taken on a Horiba SEAP-300 polarimeter (Horiba, Kyoto, Japan). UV spectra were obtained on a Hitachi UV 210A spectrophotometer (Shimadzu, Kyoto, Japan). IR spectra were measured on a Bio-Rad FTS-135 spectrometer with KBr pellets (SPECTRO, Kleve, Germany). 1D and 2D NMR spectra were recorded on a Brucker AM-400 or DRX-500 NMR spectrometer (Bruker, Karlsruher, Germany). HR-EI-MS were recorded on a Waters Auto Premier P776 spectrometer (Waters, Milford, MA, USA). Preparative HPLC was performed on an Agilent 1100 series with a Zorbax SB-C18 (5 μm, 9.4 × 150 mm) column (Agilent Technologies, Santa Clara, CA, USA). Preparative MPLC was performed on a Buchi apparatus equipped with a Buchi fraction collector C-660, a Buchi pump module C-605 and a manager C-615 (Büchi Labortechnik AG, Flawil, Switzerland). Column chromatography (CC) was performed on silica gel (200–300 mesh; Qingdao Marine Chemical Inc., Qingdao, People’s Republic of China) and Sephadex LH-20 (Amersham Biosciences, Upssala, Sweden). Fractions were monitored by TLC, and spots were visualized by heating silica gel plates sprayed with 10% H2SO4 in EtOH.
Fungal material
The fungus P. multirostrata EA-12 was isolated from fresh leaves of E. Adenophorum collected in Kunming Institute of Botany, Kunming, Yunnan province, PR China in July 2012. The fungus was identified by observing the morphological characteristics and analysis of the internal transcribed spacer regions. The result from the BLAST search indicated that the sequence was the same (100%) as that for the sequence of P. multirostrata (compared with EF585392.1). The strain is preserved at the Kunming Institute of Botany, Chinese Academy of Sciences (No. F2012076).
Fermentation, extraction and isolation
This fungal strain was cultured on potato dextrose agar medium at 25 °C for 10 days. The agar plugs were inoculated in 500-ml Erlenmeyer flasks, each containing 100 ml of potato dextrose media. Flask cultures were incubated at 28 °C on a rotary shaker at 160 r.p.m. for 2 days as seed culture. Sixty 500-ml Erlenmeyer flasks each containing 150 ml of potato dextrose broth were individually inoculated with 25 ml of seed culture and incubated at 25 °C on a rotary shaker at 160 r.p.m. for 15 days.
The fermented broth was filtered through cheesecloth to separate supernatant and mycelia. The former was extracted with EtOAc (3 × 15 liter), and the organic solvent was evaporated to dryness under vacuum to afford the extract of the supernatant. The mycelia was extracted with acetone (3 × 1 liter) and then concentrated under reduced pressure to afford an aqueous solution. The aqueous solution was extracted with EtOAc (3 × 1 liter), and the EtOAc solution was evaporated to dryness under vacuum to obtain the crude extract of the mycelia. Both extracts were combined to yield a residue (11.0 g). The residue was subjected to silica gel CC with a gradient elution system of CHCl3/MeOH (from 100:0 to 0:100, V/V) to obtain seven fractions. Fraction B was separated to six subfractions (B1∼B6) by MPLC eluted with MeOH/H2O (from 0:100 to 100:0, V/V). Fraction B2 (1.8 g) was separated by silica gel CC eluted with CHCl3/MeOH (from 100:1 to 96:4, V/V) to afford 5 (1.4 g). Fraction B3 was further separated by preparative HPLC (MeCN/H2O, from 30:70 to 60:40, V/V, 10 ml min−1 in 30 min) to yield 4 (8.0 mg, tR=16.2 min) and 6 (24.5 mg, tR=14.8 min). Fraction B5 (1.7 g) was separated by silica gel CC eluted with CHCl3/Me2CO (from 20:1 to 3:1, V/V) to obtain five subfractions (B5a∼B5e). Fraction B5b (150 mg) was further separated by preparative HPLC (MeCN/H2O, from 25:75 to 60:40, V/V, 10 ml/min in 35 min) to afford 1 (5.2 mg, tR=29.2 min), 2 (4.8 mg, tR=19.0 min) and 7 (3.1 mg, tR=25.6 min). Fraction B5c (88 mg) was subjected to sephdax LH-20 (CHCl3/MeOH, 1:1, V/V ) and silica gel CC (CHCl3/MeOH, 20:1) to yield 3 (11.2 mg).
Physico-chemical properties
Multirostratin A (1): white, amorphous solid; [α]25 D+118.0 (c 0.20 MeOH); UV (MeOH) λmax (log ɛ) 203 (4.38) nm; IR (KBr) νmax 3424, 2949, 1700, 1641, 1453, 1023, 978, 701 cm−1. 1H and 13C NMR data (see Table 1); HR-ESI-MS (pos.) m/z 445.2622 (calcd for C29H35NO3, 445.2617).
20-Oxo-deoxaphomin (2): white, amorphous solid; [α]25 D+61.1 (c 0.21 MeOH); UV (MeOH) λmax (log ɛ) 203 (4.35), 233 (3.93) nm; IR (KBr) νmax 3423, 2923, 1691, 1662, 1454, 1275, 1024, 973 cm−1. 1H and 13C NMR data (see Table 1); HR-ESI-MS (pos.) m/z 461.2566 (calcd for C29H35NO4, 461.2578).
Cytotoxic activity
Cytotoxicities of multirostratin A (1) and 20-oxo-deoxaphomin (2) were assayed against five tumor cell lines, including SK-BR-3 (breast cancer cell line), SMMC-7721 (hepatocellular carcinoma cell line), HL-60 (human myeloid leukemia cell line), PANC-1 (pancreatic cancer cell line) and A-549 (lung cancer cell line). Assays were performed as described previously.16
Supplementary
1D and 2D NMR and MS spectra of compounds 1 and 2 are available in Supplementary Information.
References
Tan, R. X. & Zou, W. X. Endophytes: a rich source of functional metabolites. Nat. Prod. Rep. 18, 448–459 (2001).
Kharwar, R. N., Mishra, A., Gond, S. K., Stierle, A. & Stierle, D. Anticancer compounds derived from fungal endophytes: their importance and future challenges. Nat. Prod. Rep. 28, 1208–1228 (2011).
Xiao, Z. E. et al. Asperterpenols A and B, new sesterterpenoids isolated from a mangrove endophytic fungus Aspergillus sp. 085242. Org. Lett. 15, 2522–2525 (2013).
Capasso, R. et al. Ascochalasin, a new cytochalasin from Ascochyta heteromorpha. J. Nat. Prod. 51, 567–571 (1988).
Aldridge, D. C., Armstrong, J. J., Speake, R. N. & Turner, W. B. The structures of cytochalasins A and B. J. Chem. Soc. C Org. 1667–1676 (1967).
Evidente, A., Andolfi, A., Vurro, M., Zonno, M. C. & Motta, A. Cytochalasins Z1, Z2 and Z3, three 24-oxa[14]cytochalasans produced by Pyrenophora semeniperda. Phytochemistry 60, 45–53 (2002).
Konig, G. M., Wright, A. D., Aust, H. J., Draeger, S. & Schulz, B. Geniculol, a new biologically active diterpene from the endophytic fungus Geniculosporium sp.1. J. Nat. Prod. 62, 155–157 (1999).
Izawa, Y., Hirose, T., Shimizu, T., Koyama, K. & Natori, S. Six new 10-pheynl-[11]cytochalasans, cytochalasins N-S from Phomopsis sp. Tetrahedron 45, 2323–2335 (1989).
Steyn, P. S., van Heerden, F. R. & Rabie, C. Cytochalasins E and K, toxic metabolites from Aspergillus clavatus. J. Chem. Soc. Perk. Trans. 1 541–544 (1982).
Liu, R. et al. 10-Phenyl-[12]-cytochalasins Z7, Z8, and Z9 from the marine-derived fungus Spicaria elegans. J. Nat. Prod. 69, 871–875 (2006).
Chen, Z. M. et al. New cytochalasins from the marine–derived fungus Xylaria sp. SCSIO 156. Helv. Chim. Acta 94, 1671–1676 (2011).
Zhang, D. W. et al. Periconiasins A-C, new cytotoxic cytochalasans with an unprecedented 9/6/5 tricyclic ring system from endophytic fungus Periconia sp. Org. Lett. 15, 1674–1677 (2013).
Pongcharoen, W., Rukachaisirikul, V., Phongpaichit, S., Rungjindamai, N. & Sakayaroj, J. Pimarane diterpene and cytochalasin derivatives from the endophytic fungus Eutypella scoparia PSU-D44. J. Nat. Prod. 69, 856–858 (2006).
Lingham, R. B. et al. L-696,474, a novel cytochalasin as an inhibitor of HIV-1 protease. III: biological activity. J. Antibiot. 45, 686–691 (1992).
Evidente, A., Andolfi, A., Vurro, M., Zonno, M. C. & Motta, A. Cytochalasins Z4, Z5, and Z6, three new 24-Oxa[14] cytochalasans produced by Phoma exigua var. heteromorpha. J. Nat. Prod. 66, 1540–1544 (2003).
Niu, S. W. et al. Lobophorins E and F, new spirotetronate antibiotics from a South China Sea-derived Streptomyces sp. SCSIO 01127. J. Antibiot. 64, 711–716 (2011).
Acknowledgements
This project was financially supported by the National Natural Science Foundation of China (U1132607, 81102346) and Youth Innovation Promotion Association of CAS (2011312D11019).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article

Cite this article
Chen, Zm., Chen, HP., Li, Y. et al. Cytochalasins from cultures of endophytic fungus Phoma multirostrata EA-12. J Antibiot 68, 23–26 (2015). https://doi.org/10.1038/ja.2014.87
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2014.87
This article is cited by
-
Fungal Endophytes: an Accessible Source of Bioactive Compounds with Potential Anticancer Activity
Applied Biochemistry and Biotechnology (2022)
-
Cytochalasins from Xylaria sp. CFL5, an Endophytic Fungus of Cephalotaxus fortunei
Natural Products and Bioprospecting (2021)
