
Abstract
New bioactive secondary metabolites, called abenquines, were found in the fermentation broth of Streptomyces sp. strain DB634, which was isolated from the soils of the Chilean highland of the Atacama Desert. They are composed of an amino acid linked to an N-acetyl-aminobenzoquinone. Isolation of the abenquines (1–4), their structure elucidation by NMR analysis and MS, as well as the kinetics of their production are presented. The abenquines show inhibitory activity against bacteria, dermatophytic fungi and phosphodiesterase type 4b. The amino acid attached to the quinone is relevant to the enzyme inhibitory activity.
Similar content being viewed by others

Introduction
The Northern Chilean highlands of the Atacama Desert comprise a number of extreme habitats, including permanent arid soils exposed to very high UV irradiation and strong fluctuations in temperature, various types of salt lakes differing in salinity and mineral salts composition and also geothermal-driven hot springs and geysers. These habitats make this area extraordinarily interesting for studies of microbial communities adapted to extreme environmental conditions. Previous studies have shown that salt lakes of this area harbor unique communities of anoxygenic phototrophic bacteria1 and cyanobacteria.2 Recently, we have isolated a number of actinobacteria from arid soils of the Atacama Desert and characterized them in regard to their potential to synthesize secondary metabolites. In this study, we present the secondary metabolite profile of one of the isolated Streptomyces strains.
Materials and methods
Producing organism and taxonomy
For isolation of the strain DB634, a soil sample taken at Salar de Tara (4500 m above sea level) was air-dried at 50 °C. The dry soil sample was pre-incubated in yeast extract medium at 50 °C for 1 h and then transferred to malt extract yeast extract glucose agar (ISP2) supplemented with 50 μg ml−1 cycloheximide (Sigma-Aldrich, Steinheim, Germany). A colony of the strain DB634 was picked from such an agar plate after 11 days of incubation at 28 °C.
For taxonomical characterization the DNA extraction was performed using the QIAGEN DNeasy Tissue Kit (QIAGEN, Hilden, Germany). The digestion of the bacterial cell wall was performed with an enzymatic lysis buffer, containing 20 mg ml−1 lysozyme (SIGMA Life Science, München, Germany). For amplification of the 16S rRNA gene, the universal eubacterial primers 27F and 1492R3 (MWG Biotech AG, Ebersberg, Germany) and the DreamTaq Green PCR Master Mix (Fermentas, St Leon-Rot, Germany) were used. The resulting sequences were compared with sequences in the EMBL nucleotide database available online at the European Bioinformatics Institute homepage using the Basic Local Alignment Search Tool (nucleotide BLAST) and of RDP-II Project homepage.
Air-dried samples of aerial mycelium from strain DB634 with spore chains were sputter coated with gold/palladium, and micrographs were taken with a Zeiss DSM940 scanning electron microscope (Zeiss, Jena, Germany).
Fermentation and isolation
For isolation of the abenquines, strain DB634 was grown in a medium that consists of starch 10 g, peptone from soymeal (Merck Bioscience, Darmstadt, Germany) 5 g and artificial sea salt (Tropic Marin, Dr Biener GmbH, Wartenberg, Germany) 10 g l−1 deionized water called SPS medium. In some experiments, this medium was supplemented with 1 g of phenylalanine or tryptophane per liter medium. For measuring the fermentation kinetics, the strain was cultivated in 100 ml of SPS medium in 300-ml shaking flasks with four baffles. For production of the abenquines, the strain was cultivated in 10 times 1 l of SPS medium with and without supplementation with amino acids, in 2-l shaking flasks with a single baffle. All fermentations were carried out for 9–11 days on a rotary shaker at 120 r.p.m. and 28 °C.
For isolation of 1, 2a/b and 3, a 10-l fermentation in SPS medium supplemented with phenylalanine was harvested. The fermentation broth was separated by centrifugation into culture filtrate and mycelium. The filtrate was applied on an Amberlite XAD-16 column and eluted with H2O-EtOH (9:1) as well as H2O-EtOH (6:4). The fractions containing 1, 2a/b and 3 were concentrated in vacuo, the aqueous residue was acidified with HCl to pH 4 and extracted with EtOAc. The organic extracts were concentrated in vacuo, to yield crude extracts of 1.6 g and 1.3 g. These extracts were fractionated by column chromatography on Sephadex LH-20 (GE Healthcare, Uppsala, Sweden, 3 × 80 cm, MeOH). Fractions containing 1, 2a/b and 3 were pooled and further purified by preparative RP-HPLC (Phenomenex Gemini C18 110A AXIA, 100 × 21.20 mm, Phenomenex, Aschaffenburg, Germany) with CH3CN–0.1% HCOOH (aq) as solvents using linear gradient elution from 25 to 45% CH3CN over 15 min at a flow rate of 15 ml min−1 and UV detection at 320 nm. This yielded 362 mg of 1, 20 mg of 2a/b (in a ratio of approximately two-thirds of 2a and one-third of 2b) and 11 mg of 3.
For isolation of 4, 10 l of SPS medium supplemented with tryptophane was used for fermentation. The isolation of 4 was carried out according to the above-described isolation of 1, 2a/b and 3. This yielded 21 mg of 4.
Production kinetics
To measure the abenquine A and phenylalanine concentrations in the fermentation broth, samples were aseptically taken, filtered and analyzed by reversed-phase HPLC-UV/MS (for details see General Experimental Procedures section). For detection of the abenquines, the standard gradient was applied (0 min 5% B, 4 min 60% B, 6 min 100% B; flow 2 ml min−1), whereas for phenylalanine an optimized gradient (0 min 0% B, 4 min 0% B, 6 min 100% B; flow 2 ml min−1) was applied. The abenquines were detected by UV absorbance at 322 nm and phenylalanine was detected by mass spectroscopy (166 m/z). For quantification of the compounds standard solutions were analyzed.
To determine the starch concentration in the fermentation broth, 5 μl of a sample was diluted with 190 μl H2O and dyed with 10 μl iodine-potassium iodide solution in a 96-well plate. Absorbance was measured using the microplate reader Infinite M200 (Tecan, Mannedorf, Switzerland) at excitation 565 nm. For quantification, standard solutions with defined concentrations were analyzed.
Antimicrobial assays
Antimicrobial assays were performed using Bacillus subtilis DSM 347, Staphylococcus lentus DSM 6672, Xanthomonas campestris DSM 2405 and Candida albicans DSM 1386. Overnight cultures of the test organisms in tryptic soy broth were diluted to an OD (600 nm) of 0.01–0.05. The assays were prepared by transferring 2 μl of a 10 mM solution of the sample compound dissolved in DMSO into one well of a 96-well microtiter plate containing 200 μl of cell suspension culture. After the microtiter plates were incubated for 5 h at 37 °C (B. subtilis, St. lentus and C. albicans) and 14–16 h at 28 °C (X. campestris), 10 μl of a resazurin solution (0.2 mg ml−1 phosphate-buffered saline) was added to each well and the plates were incubated again for 5–30 min. To evaluate cell viability, the reduction of resazurin to resorufin was assessed by measuring the absorbance 600 nm (reference 690 nm). The resulting values were compared with a positive (10 μM chloramphenicol for bacteria; 10 μM Nystatin for the yeast) and a negative control (no compound) on the same plate.
Cytotoxicity assays
The sensitivity of mouse fibroblasts (NIH-3T3 cell line kindly provided by G Rimbach, University of Kiel, Germany) to the abenquines was evaluated by monitoring the metabolic activity using the CellTiter-BlueCell Viability Assay (Promega, Mannheim, Germany). NIH-3T3 cells were maintained in RPMI 1640 medium. Media were supplemented with 10% fetal bovine serum (Promocell, Heidelberg, Germany), 100 IU ml−1 penicillin and 100 μg ml−1 streptomycin (Promocell). The cultures were maintained at 37 °C under a humidified atmosphere and 5% CO2. The cell lines were transferred every 3 or 4 days. For experimental procedures, cells were seeded in 96-well plates at concentrations of 7500 cells per well. After 24-h incubation, the medium was removed and 100 μl of fresh medium with the test sample dissolved inside was added to the cells. Each sample was prepared in triplicate. Tamoxifen, as a standard therapeutic drug, was used as a positive control. Following compound addition, plates were cultured for 24 h at 37 °C. Afterwards, the assay was performed according to the manufacturer's instructions. Cells were incubated for 2 h at 37 °C. Fluorescence was measured using the microplate reader Infinite M200 (Tecan) at excitation 560 nm and emission 590 nm.
PDE4 assays
Analysis of effects on human recombinant cyclic AMP (cAMP)-specific phosphodiesterase (PDE-4B2) was carried out with the diluted test sample in 30 μl of 50 mM Tris–HCl buffer (pH 7.5) containing 8.3 mM MgCl2, 1.7 mM EGTA and 5 U recombinant human cAMP-specific PDE-4B2 (Biocat 60042-BPS, Heidelberg, Germany). The reaction was started with 10 μl of 12 μM cAMP (Sigma A9501, Taufkirchen, Germany) dissolved in 50 mM Tris–HCl buffer (pH 7.5) containing 8.3 mM MgCl2, 1.7 mM EGTA.
After an incubation period of 30 min at 30 °C, the reaction was stopped and the adenosinemonophosphate concentration was quantified with the PDELight HTS cAMP Phosphodiesterase Assay Kit (Lonza, LT07-600, Wuppertal, Germany), according to the instructions of the supplier. The luminescence was measured using the microtiter plate reader Infinite M200 (Tecan) with 0.1 s integration time.
For inhibition of the PDE, 1 μM and 10 μM of rolipram (4-[3-(cyclopentyloxy)-4-methoxyphenyl]-2-pyrrolidinone; Merck Bioscience, cat. no. 557330) were used as positive controls.
Antifungal assays
Tested compounds were applied on an assay disc (Ø 6 mm) and the disc then placed to the center of an agar plate previously inoculated with fungal spores. For inoculation, 1 × 105 spores from three different dermatophytes (Trichophyton rubrum, T. mentagrophytes, Microsporum canis) were dispensed with a sterile spatula on an agar plate containing Sabouraud medium (peptone 10 g, glucose 20 g and agar 15 g in 1.0 l tap water). After incubation for 6 days at 28 °C, the diameter of inhibition zones were measured.
Preparation and analysis of D- and L-FDVA derivatives
Compounds 1–4 (2 mg each) were hydrolyzed by heating in HCl (6 M; 1 ml) for 5 h at 100 °C. The solutions were then evaporated to dryness and redissolved in H2O (250 μl). A 1% (w/v) solution (100 μl) of D-FDVA [Nα-(2,4-dinitro-5-fluorophenyl)-D-valinamide] in acetone was added. After addition of NaHCO3 solution (1 M; 40 μl) and DMSO (70 μl), the mixture was incubated at 60 °C for 2 h. The reactions were quenched by addition of HCl (2 M; 30 μl). The reaction mixtures were then diluted 100-fold with MeOH–H2O (1:1) and analyzed by analytical HPLC (see General experimental procedures section). The amino-acid standards were derivatized with both D- and L-FDVA. Retention times (min) of the amino-acid derivatives were as follows: D-FDVA-L-Leu (4.1), L-FDVA-L-Leu (3.5), D-FDVA-L-Phe (4.1), L-FDVA-L-Phe (3.8), D-FDVA-L-Val (3.8) and L-FDVA-L-Val (3.2). HPLC analyses of the hydrolysates of 1, 2a/b and 3 derivatized with D-FDVA showed peaks at 4.1 min for 1, at 4.1 min for 2 and at 3.8 min for 3.
Compound 4 was hydrolyzed with HCl (6 M; 400 μl; 100 °C; 1 h) containing phenol (3% w/v). The hydrolysate was derivatized with D-FDVA as described above. For the HPLC analysis of the tryptophan derivatives, the HPLC conditions were the same as above. The retention times of the derivatized amino-acid standards were as follows: L-FDVA-L-Trp (3.5) and D-FDVA-L-Trp (4.1). The derivatized hydrolysate showed a peak corresponding to L-Trp.
General experimental procedures
The optical rotation was measured on a Perkin Elmer model 241 polarimeter (Perkin Elmer, Waltham, MA, USA). UV spectra were obtained on a NanoVue (GE Healthcare) photometer. NMR spectra were recorded on a Bruker DRX500 spectrometer (Bruker Daltonics, Bremen, Germany, 500 and 125 MHz for 1H and 13C NMR, respectively), using the signals of the residual solvent protons and the solvent carbons as internal references (δH 2.04 and δC 29.8 p.p.m. for acetone-d6). High-resolution mass spectra were acquired on a benchtop time-of-flight spectrometer (MicrOTOF II, Bruker Daltonics) with positive ESI. Analytical reversed-phase HPLC-UV/MS experiments were performed using a C18 column (Phenomenex Onyx Monolithic C18, 100 × 3.00 mm, Phenomenex, Aschaffenburg, Germany) applying an H2O (A)/CH3CN (B) gradient with 0.1% HCOOH added to both solvents (gradient: 0 min 5% B, 4 min 60% B, 6 min 100% B; flow 2 ml min−1) on a VWR Hitachi Elite LaChrom system coupled to an ESI-ion trap detector (Esquire 4000, Bruker Daltonics).
Preparative HPLC was carried out using either a LaPrep HPLC system (VWR International, Darmstadt, Germany) with a P110 pump, a P311 UV detector, a Labocol vario-200 fraction collector, a Smartline 3900 autosampler and a Phenomenex Gemini-NX 10 m C18 110 A AX, 100 × 50 mm column or a VWR Hitachi (VWR International) system consisting of an L-7150 pump, an L-2200 autosampler and an L-2450 diode array detector and a Phenomenex Gemini C18 110A AXIA, 100 × 21.20 mm column.
Results
Isolation and identification of the producing strain
Strain DB634 was isolated from a soil sample taken at Salar de Tara (4500 m above sea level). The morphological characteristics and genetic sequence information of the 16S rRNA gene designated the strain as belonging to the genus Streptomyces.4 The substrate mycelium of the bacterium is beige, the aerial mycelium is white and monopodially branched with straight, long aerial hyphae. The sporogenous hyphae form primitive spirals and the conidia are of the spiny spore type (Figure 1). The spores appear dark-grey in color.

Spore chain of Streptomyces sp. strain DB634 (scanning electron microscopic photograph).
The most closely related strain type according to the 16S rRNA gene sequence (1380 bp) is Streptomyces caelestis DSM 40084, with a sequence similarity of 100% (EMBL nucleotide database).
Fermentation and isolation of the abenquines
For production of 1, the strain DB634 was cultivated in the complex SPS medium. In the first 5 days, the bacteria grew by consumption of the starch and degradation of peptone. Owing to the release of amino acids from the peptone, the phenylalanine concentration in the medium rose from an initial level of 11 mg l−1 of free phenylalanine up to 52 mg l−1 after 6 days (Figure 2). The production of 1 started after 3 days and rose to a maximal yield of 11 mg l−1 after 6 days. The concentrations of both 1 and phenylalanine decreased after 6 days (Figure 2). Although there was no limitation of phenylalanine in the media during the production of 1, increasing the phenylalanine concentration in the media also increased the production of 1. In the SPS medium, supplemented with 1 g l−1 phenylalanine, a maximal production of 1 was achieved, with a yield of 138 mg l−1 after 6 days.

Kinetics of starch (-▴-), phenylalanine (-▪-) and abenquine A (-•-) concentrations during the fermentation process (mean values of four independent fermentations, for standard deviations see supplementary information).
In order to analyze the possible incorporation of other amino acids into the compounds produced, we supplemented the SPS medium with 1 g l−1 of tryptophan. Interestingly, this led to the production of high concentrations of 4.
For isolation of 1, 2a/b and 3, strain DB634 was cultivated in 10 l of SPS medium with and without phenylalanine supplementation. For isolation of 4, 10 l of SPS medium supplemented with 1 g l−1 tryptophan was used. After harvesting, the abenquines were purified from the supernatant by chromatography on Amberlite XAD-16, Sephadex LH-20 and RP-HPLC (for further details see Materials and methods section). Fermentation in 10 l of SPS medium without supplementation gave a yield of 12 mg of 1. Fermentation in 10 l of SPS medium with 1 g l−1 phenylalanine supplementation gave a yield of 362 mg of 1. From the extract of this fermentation we could also isolate 20 mg of 2a/b and 10 mg of 3. The supplementation with 1 g l−1 tryptophan gave a yield of 21 mg of 4.
Structure elucidation
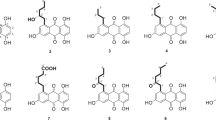
The structure elucidation of the compounds (Figure 3) was mainly based on the interpretation of 1D- and 2D-NMR spectra of 1 (Table 1 and Supplementary information). The molecular formula C17H16N2O5 was determined by HR-MS, high-resolution ESI-mass spectra, and was in full agreement with the delineated structure. The structure was composed of three different parts. The first one was a quinone core structure, which consisted of two ketone carbonyl groups, C-1 (δC 179.6) and C-4 (δC 183.9), two nitrogen-bearing olefinic carbons, C-2 (δC 142.4) and C-5 (δC 147.5) and two sp2 hybridized methines, CH-3 (δH 7.27, δC 109.7) and CH-6 (δH 5.45, δC 95.9). The signals of H-3 and H-6 formed a singlet each, which already suggested the para-orientation of the two hydrogen atoms. Further proof was given by the 1H-13C HMBC correlations (Figure 4): the signal of H-3 showed very strong correlations to the resonances of C-1 and C-5, whereas H-6 showed intense long-range couplings to C-2 and C-4. The second substructure was an N-acetyl residue located at C-2. The shifts of CH3-2′ (δH 2.24, δC 24.7) had values rather characteristic of methyl groups belonging to acetyl residues. The signal of H3-2′ appeared as a singlet with a long-range coupling to C-1′ (δC 170.9) as its only correlation. C-2 as the site of the N-acetylation became clear after the third part of the molecule, the phenylalanine residue, was delineated from the spectroscopic data. The phenylalanine was clearly shown to be located at C-5 due to an 1H-13C HMBC correlation from the resonance of the amino acid α-CH, H-2′′ (δH 4.52) to C-5. The signal of H-2′′ showed 1H-1H COSY correlations to the resonances of H2-3′′ (δHa 3.36, δHb 3.25) and also showed the corresponding splitting pattern. H2-3′′ showed long-range couplings to the resonance of C-1′′ (δC 171.6), the acid carbonyl carbon, as well as to carbons belonging to the aromatic part of the phenylalanine residue, that is, C-4′′ (δC 137.2) and C-5′′/9′′ (δC 130.2). The aromatic protons H-5′′ (δH 7.26) through to C-9′′ (δH 7.26) formed a coherent spin system, which completed the last substructure of the compound. Thus, the planar structure of the molecule was determined. The structures of abenquines B–D were determined after high-resolution mass measurements and analysis of 1H- and 13C-NMR data (see Tables 2 and 3).

Chemical structures of the abenquines.

Selected HMBC correlations for 1.
Neither the quinone nor the acetyl moiety contained asymmetric carbons, thus the respective amino acids were the only optically active parts of the molecules. To determine the absolute configuration, 1–4 were hydrolyzed, derivatized with D-FDVA and analyzed by HPLC-MS in comparison with standards. All amino acids, that is, the respective leucyl, phenylalanyl, tryptophyl and valyl residues of the compounds were present in L-configuration.
Biological activity
Abenquine A (1) and abenquine D (4) showed moderate inhibitory activity against the phosphodiesterase type 4 (PDE4b). The activities of abenquine B1/B2 (2a/b) and abenquine C (3) were significantly lower (Table 4). For comparison of the inhibitory activity we used the PDE type 4 standard inhibitor rolipram.5
The abenquines showed poor activity against B. subtilis (Table 4) and no activity against S. lentus, X. campestris and C. albicans. The antiproliferative activity against mouse fibroblasts (NIH-3T3 cell line) was weak as well (Table 4).
Abenquine A (1) showed poor antifungal activity against dermatophytic fungi (T. rubrum, T. mentagrophytes, M. canis, for further details see supplementary information).
Discussion
Streptomyces sp. strain DB634 was taxonomically identified as belonging to the genus Streptomyces. An unambiguous identification on the species level was not possible. According to 16S rRNA gene sequence similarity the strain is closely related to S. caelestis. However, the spore surface of strain DB634 clearly showed them to be spiny-type spores, which disagrees with the description of rugose-type spores for S. caelestis.6
The abenquines are simple aminobenzoquinones substituted with different amino acids. Even though aminobenzoquinones are often found in natural products, the substitution with an unmodified amino acid is rare. Structurally related compounds that share N-substituted aminobenzoquinones as a structural feature are lepiotaquinone (from Lepiota americana)7 and lilacinone (from Lactarius lilacinus),8 which are found as pigments in fungi. Among bacterial metabolites, the aminobenzoquinone substructure is found, for example, in ansamycin-type antibiotics or mitomycin.9 Interestingly, apart from fungi and bacteria, sponges also produce metabolites such as nakijiquinones,10 which emerge from benzoquinones and amino acids.
The fact that among natural products the abenquines represent rare combinations of benzoquinones and amino acids certainly raises questions concerning their biosynthesis. The reaction of quinones with amino acids has been reported to take place under mild conditions and without the involvement of enzymes.11 The chemical reaction of amino acids with benzoquinone has even been described as a method for the quantitative determination of amino acids.10 Thus, a produced N-acetyl-aminobenzoquinone might function as a Michael acceptor for a number of amino acids. The wide spectrum of amino acids that are incorporated in the course of our feeding experiments supports this assumption. However, the possibility that one or more enzymes are involved in the formation of the abenquines cannot be ruled out. Especially, the very short production time and the rapid degradation of the abenquines suggest the involvement of enzymes. A laccase could be a possible candidate responsible for the formation of the abenquines, because enzymes of this group have been reported to mediate the adduction of amino acids to hydroquinones.12
The abenquines inhibit PDE4. In comparison with the selective PDE4 inhibitor rolipram,5 the abenquines exhibited fivefold less potent activity. PDE4 degrades the ubiquitous second messenger cAMP to adenosinemonophosphate. Therefore, selective PDE4 inhibitors have become an important pharmacological tool as novel therapeutics for, for example, the treatment of inflammatory diseases such as asthma, chronic obstructive pulmonary disease or multiple sclerosis.13 Interestingly, we could already show that the amino-acid residue of the molecule is of importance for the activity, that is, the two compounds with aromatic amino acids have a much more potent activity than the molecules comprising the two smaller and non-aromatic ones.
References
Thiel, V. et al. Unique communities of anoxygenic phototrophic bacteria in saline lakes of Salar de Atacama (Chile): evidence for a new phylogenetic lineage of phototrophic Gammaproteobacteria from pufLM gene analyses. FEMS Microbiol. Ecol. 74, 510–522 (2010).
Dorador, C., Vila, I., Imhoff, J. F. & Witzel, K. P. Cyanobacterial diversity in Salar de Huasco, a high altitude saline wetland in northern Chile: an example of geographical dispersion? FEMS Microbiol. Ecol. 64, 419–432 (2008).
Lane, D. L. 16S/23S rRNA sequencing. Nucleic acid techniques In Bacterial Systematics, (Stackebrandt, E. & Goodfellow, M., eds) pp. 115–175 (John Wiley & Sons, New York, 1991).
Williams, S. T., Goodfellow, M. & Alderson, G. Genus Streptomyces Waksman and Henrici 1943, 339AL In Bergey's Manual of Systematic Bacteriology Vol. 4 (eds Williams, S.T. et al.) pp. 2452–3492 (Williams & Wilkins, Baltimore, 1989).
Kanes, S. J. et al. Rolipram: a specific phosphodiesterase 4 inhibitor with potential antipsychotic activity. Neuroscience 144, 239–46 (2007).
Wink, J. M. Compendium of Actinobacteria, University of Braunschweig. Copyright Dr Joachim M. Wink, Sanofi-Aventis Deutschland GmbH, Höchst.
Aulinger, K., Arnold, N. & Steglich, W. Metabolites of 2-aminophenol from fruit bodies of Lepiota americana (Agaricales). Z. Naturforsch 55 c, 481–484 (2000).
Spiteller, P., Arnold, N., Spiteller, M. & Steglich, W. Lilacinone, a red aminobenzoquinone pigment from Lactarius lilacinus. J. Nat. Prod. 66, 1402–1403 (2003).
Floss, H. G., Yu, T. W. & Arakawa, K. The biosynthesis of 3-amino-5-hydroxybenzoic acid (AHBA), the precursor of mC7N units in ansamycin and mitomycin antibiotics: a review. J. Antibiot. 64, 35–44 (2011).
Shigemori, H., Madono, T., Sasaki, T., Mikami, Y. & Kobayashi, J. Nakijiquinones A and B, new antifungal sesquiterpenoid quinones with an amino acid residue from an Okinawan marine sponge. Tetrahedron 50, 8347–8354 (1994).
Morrison, M., Steele, W. & Danner, D. J. The reaction of benzoquinone with amines and proteins. Arch Biochem. Biophys. 134, 515–523 (1969).
Wellington, K. W., Steenkamp, P. & Brady, D. Diamination by N-coupling using a commercial laccase. Bioorgan. Med. Chem. 18, 1406–1414 (2010).
Houslay, M. D., Schafer, P. & Zhang, K. Y. J. Phosphodiesterase-4 as a therapeutic target. Drug Discov. Today 10, 1503–1519 (2005).
Acknowledgements
We gratefully thank R Schmaljohann for microscopy, G Kohlmeyer-Yilmaz, M Höftmann, as well as Dr F Sönnichsen for running and processing NMR experiments. This study is from the Kieler Wirkstoff-Zentrum, which is supported by the Ministry of Science, Economic Affaires and Transport of the State of Schleswig-Holstein (Germany), in the frame of the ‘Future Program for Economy’, which is co-financed by the European Union (EFRE).
Author information
Authors and Affiliations
Corresponding author
Additional information
Dedicated to Professor Gerhard Bringmann on the occasion of his 60th birthday.
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article
Cite this article
Schulz, D., Beese, P., Ohlendorf, B. et al. Abenquines A–D: aminoquinone derivatives produced by Streptomyces sp. strain DB634. J Antibiot 64, 763–768 (2011). https://doi.org/10.1038/ja.2011.87
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2011.87
Keywords
This article is cited by
-
Asenjonamides A–C, antibacterial metabolites isolated from Streptomyces asenjonii strain KNN 42.f from an extreme-hyper arid Atacama Desert soil
The Journal of Antibiotics (2018)
-
Natural product diversity of actinobacteria in the Atacama Desert
Antonie van Leeuwenhoek (2018)
-
Isolation and anti-HIV-1 integrase activity of lentzeosides A–F from extremotolerant lentzea sp. H45, a strain isolated from a high-altitude Atacama Desert soil
The Journal of Antibiotics (2017)
-
Acetylcholinesterase Inhibitors from a Marine Fungus Talaromyces sp. Strain LF458
Marine Biotechnology (2015)
-
Streptomyces leeuwenhoekii sp. nov., the producer of chaxalactins and chaxamycins, forms a distinct branch in Streptomyces gene trees
Antonie van Leeuwenhoek (2014)
