
Abstract
Romidepsin (Istodax), a selective inhibitor of histone deacetylases (HDACs), was approved for the treatment of cutaneous T-cell lymphoma in November 2009 by the US Food and Drug Administration. This unique natural product was discovered from cultures of Chromobacterium violaceum, a Gram-negative bacterium isolated from a Japanese soil sample. This bicyclic compound acts as a prodrug, its disulfide bridge being reduced by glutathione on uptake into the cell, allowing the free thiol groups to interact with Zn ions in the active site of class I and II HDAC enzymes. Due to the synthetic complexity of the compound, as well as the low yield from the producing organism, analogs are sought to create synthetically accessible alternatives. As a T-cell lymphoma drug, romidepsin offers a valuable new treatment for diseases with few effective therapies.
Similar content being viewed by others


Introduction to Histone Deacetylase (HDAC) Inhibitors
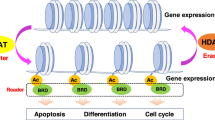
Alteration of gene expression because of epigenetic modification of the chromatin structure has been implicated as an important factor in tumorigenesis.1, 2 In chromatin, DNA is tightly coiled around core histone proteins, forming nucleosomes; linker histones bind nucleosomes together as chromatin. Methylation, acetylation, phosphorylation, ADP-ribosylation or ubiquitination of the histone tail regions affect the binding of the histone proteins to DNA, changing the structure of the chromatin and subsequently the accessibility of transcription factors to DNA regions.3 Such epigenetic processes alter levels of gene expression without changing the nucleotide sequence.4 Of these modifications, histone acetylation is the most widely studied, because of its role in the development and progression of tumors.2
Histone acetylation is controlled by histone acetyltransferases and histone deacetylases (HDACs). Histone acetyltransferases direct the addition of acetyl groups to lysine residues in the amino-terminal histone tails, neutralizing that portion of the protein and relaxing the chromatin structure, thereby increasing accessibility of transcription complexes as well as recruiting transcription cofactors.2, 3, 5, 6 Conversely, HDACs remove acetyl groups from the histone tails, resulting in a more compact and inaccessible form of chromatin, consequently silencing transcription.2, 3, 5 These effects are highly localized, and only 2–5% of encoding genes are transcriptionally regulated by the histone acetylation state.2 Importantly, this selection includes genes that control the cell cycle and apoptosis, and HDACs have been associated with several well-characterized oncogenes and tumor-suppressor genes.7, 8 Certain tumors have overexpressed HDACs and downregulated or mutated histone acetyltransferases.9 It has been suggested that an imbalance of HDAC relative to histone acetyltransferase activity can lead to a diminished expression of such regulatory genes and ensuing tumorigenesis.9
HDAC inhibitors have been found to have therapeutic properties in many different human tumor cell lines, including those derived from the bladder, breast, prostate, lung, ovary and colon, among others,10 indicating that the inhibition of HDAC activity may be a viable strategy for the treatment of cancers.11, 12, 13 It is also likely that HDAC inhibitors affect a number of subcellular pathways, and time will tell what impact these other mechanisms have on treatment of cancer and/or other diseases. HDAC inhibitors can be divided into four classes, namely hydroximates, cyclic peptides, aliphatic acids and benzamides.14 A new HDAC inhibitor, romidepsin, which has been referred to by several different names in the literature, including FK228, FR 901228, NSC 630176, depsipeptide and Istodax, is a pentapeptide isolated from cultures of Chromobacterium violaceum.15 The bicyclic depsipeptide structure of romidepsin (1, Figure 1) is composed of four amino acids (D-valine, D-cysteine, Z-dehydrobutyrine, L-valine) and (3S,4E)-3-hydroxy-7-mercapto-4-heptenoic acid, and the numbering of 1 is based on Xiao et al.16 (Note that the structures of romidepsin (1) and its reduced counterpart (2) were published incorrectly in the review by Grant et al.17) Romidepsin was approved by the US Food and Drug Administration (FDA) in 2009 for use in patients with cutaneous T-cell lymphoma (CTCL).

Background on CTCL
The incidence of CTCL in the general population is estimated to be 1–4/100 000,18, 19 with approximately 1500 new cases and 500 deaths per year in the United States.20 CTCL is twice as prevalent in males as in females, and the median age of presentation is 57 years. In the United States, the disease is also more common among African Americans than in other race categories.19 Approximately, 16 000–20 000 individuals in the United States are affected by CTCL.20 One study from 1973 to 1984 found a 3.2-fold increase in the incidence of CTCL over that time period;21 a similar study from 1973 to 2002 reported a 3.4-fold increase in incidence over the course of the study.22
The most common types of CTCL are mycosis fungoides (MF), so called because early physicians believed it to be a fungal infection, although it is not, and Sézary syndrome. The latter, the leukemic variant, is characterized by pruritus, generalized exfoliative erythroderma and abnormal lymphoid cells in the blood. On the other hand, the blood is usually not affected in MF, especially in the early stages. Skin lesions are characterized as patches, plaques or tumors that have a ‘bathing trunk’ distribution. The lymphoma may extend to cover a large surface area, lymph nodes, and, in later stages, viscera.
MF is the most common type of CTCL, and is characterized by pink or erythematous scaly patches and plaques, with varying degrees of scaling and pruritus.23 At the time of diagnosis, the majority of patients with MF have limited plaques on ⩽10% of their total body surface. However, approximately one-third of them have extensive plaques. Relatively small proportions of patients present with cutaneous tumors (16%) or generalized erythroderma (12%). Non-skin lesions are more likely to be seen in later stage disease, with 8% seen in stage T2, 30% in stage T3 and 42% in stage T4. The primary non-skin and non-lymph node sites are lung, gastrointestinal, liver and central nervous system. Bone marrow involvement is rare in early stage disease.
CTCL is uniformly fatal. The ultimate cause of death for patients with CTCL is commonly infection, particularly sepsis, from Pseudomonas aeruginosa or Staphylococcus aureus, caused by chronic skin infection and subsequent systemic infections.24 Prognosis is dependent on age as well as disease stage at diagnosis,25 with earlier stage disease associated with higher 5-year overall survival and disease-free survival rates than later stage disease. The overall 5-year survival rate for patients with MF is 91%, while it is only 40% for those with Sézary syndrome.26 Typically, those presenting with early stages of CTCL have a favorable prognosis, with around 95% 5-year survival;25, 27, 28 more advanced stages have lower survival rates ranging from 30 to 84%,25, 27, 28 again depending on form and stage of the cancer. According to one study, the 10-year survival rate drops from around 100% for those with stage T1 CTCL to 67% for T2; by stage T3, this number drops to 40%.29 Although most patients present with the early-stage disease, approximately 10–20% of these will progress to a later and often fatal disease.25, 27, 30 Patients that undergo transformation to large-cell lymphoma have a mean survival ranging from 2 to 19 months.30
Discovery and development of romidepsin
Romidepsin was discovered in the early 1990s through a program that was evaluating fermentation products for antimicrobial and antitumor activities. The producing organism, C. violaceum, is a Gram-negative, rod-shaped bacterium with a single polar flagellum. The strain was isolated from a soil sample collected in the Yamagata-prefecture, Japan.15 Presently, the commercial supply of romidepsin is generated via fermentation, although the yield and exact conditions are proprietary.
C. violaceum was cultured in nutrient broth with additional glucose and incubated at 30 °C aerobically with agitation. The organism reached stationary phase after 72 h, providing a maximal yield of romidepsin at this time of 19 μg ml–1, which was present in the aqueous part of the culture. The fermentation was filtered, and the filtrate extracted twice with EtOAc. The extract was evaporated in vacuo to give an oily residue, which was then purified via normal phase chromatography to yield a yellow powder.
This powder was dissolved in CH2Cl2:CH3OH:CH3CN (10:1:20), and romidepsin crystallized as colorless prisms. It displayed weak antifungal but no antibacterial activity, but it showed potent cytotoxicity against several human lung carcinoma cell lines, as well as human stomach, breast and colon adenocarcinoma cell lines.15 The structure of romidepsin (1), which is a cage-shaped bicyclic depsipeptide, was determined using a combination of spectroscopic techniques, primarily NMR and X-ray crystallography.31
Romidepsin demonstrated the ability to reverse the effects of the ras oncogene in vitro, which has been shown to have a role in tumor development; expression of the Ha-ras oncogene appears to be directly correlated with tumorigenic potential.32 As a result of romidepsin's ability to reverse the ras-transformed phenotype to normal, as well as its cytotoxic effects, it was developed initially by the US National Cancer Institute (NCI) as an anti-ras compound,33, 34 but recently it has been shown to be an HDAC inhibitor.35, 36 A program screening a number of known microbial metabolites for transcriptional activation of the SV40 promoter identified romidepsin as an antitumor compound.35 Comparison of its activity to that of trichostatin A (a known HDAC inhibitor) revealed that romidepsin was a new HDAC inhibitor.35
Defining a clear path for the development of romidepsin was complicated somewhat because of the many different parties involved. Fujisawa Corporation (now Astellas) discovered the compound in the early 1990s via a search for molecules that would revert the ras-driven oncogenic phenotype, and then, under a Cooperative Research and Development Agreement, further oncology investigations were initiated by the US NCI in 1998. When the NCI confirmed that romidepsin was a powerful anticancer agent, Fujisawa Corporation initiated their own, separate clinical trials program in 2002. Romidepsin was licensed to Gloucester Pharmaceuticals in 2004; the company was later acquired by Celgene Corporation (Summit, NJ, USA) in 2010. Gloucester approached the US FDA (beginning in 2004), obtained both Orphan Drug Status and Fast Track Status, and were able to come to an agreement, through the FDA Special Protocol Assessment process, on a path to approval, which was a pivotal single arm (phase II) design, although data from the NCI's ongoing study was also to be part of the new drug application filing. Essentially, the activity of romidepsin, coupled with the unmet medical need in CTCL, were persuasive enough arguments to move the drug forward.
Mechanisms of action
Romidepsin is converted in cells to its active form by the reduction of the disulfide bond by glutathione, resulting in a monocyclic dithiol (2, Figure 1),16, 37 and thus romidepsin serves as a prodrug that is activated only after uptake into cells. This reduced form of romidepsin (2) is inactivated rapidly in serum, perhaps due to sequestration by serum proteins.37 A cysteine in the active site pocket (Cys-151 in HDAC1) was thought to covalently bind with the reduced sulfur atom at position 13, but a mutant HDAC1 in which Cys-151 was replaced by a serine was still sensitive to romidepsin, though a higher concentration (∼eightfold) of the drug was necessary for inhibition.37 This fact, combined with the reversibility of the inhibition, suggests that while the cysteine may have a role in the affinity of the drug, it most likely does not bind covalently to the sulfur atom.37 Crystallographic and computer modeling studies using a HDAC-trichostatin A complex suggested that one of the thiol groups of the reduced depsipeptide interacts with zinc ions in the active site pocket of certain enzymes, preventing access of the substrate.38 A computer modeling study by Furumai et al.37 concluded that when interacting with HDAC1 the sulfur atom of the reduced romidepsin was located at a position that allowed interaction with the zinc ion via a water molecule. They also suggested that, based on a similar mechanism in protease inhibitors, the sulfur may tetrahedrally coordinate with the zinc, displacing the water molecule.37 The interaction between the thiol group and the active site of the enzyme prevents other substrates from binding.
The reduced form more strongly inhibits HDAC1 and HDAC2 enzymes (class I) than HDAC4 and HDAC6 enzymes (class II).37 By inhibiting class I HDAC, romidepsin inhibits the removal of acetyl groups from the lysine residues of N-terminal histone tails, maintaining a more open and transcriptionally active chromatin state.5 In addition, romidepsin also results in altered acetylation of other nuclear and cytoplasmic proteins, although the precise pathways by which it affects the cell cycle, apoptosis and angiogenesis have not been defined completely.39
Cell cycle arrest
The induction of growth arrest and/or apoptosis by romidepsin depends on the cell line tested and the concentration of drug applied.39 Growth arrest, as opposed to apoptosis, is the predominant response in cell lines in which the application of romidepsin induces the expression of the p21 tumor-suppressor gene.40 Cell lines with reduced p21 expression preferentially undergo apoptosis.41 Tumor cells are more susceptible to romidepsin than normal cells,15 a fact that leads to a viable therapeutic index in the clinic.
Direct acetylation of non-histone proteins may also trigger growth arrest or apoptosis, and several pathways have been studied, although the complete role of HDAC inhibitors is not fully understood. Exposure of whole cells to romidepsin results in a decrease in cyclin D1 and c-Myc, accompanied by an increase in p53-independent p21 WAF1/Cip 1 induction. The p21 induction leads to inhibition of cyclin-dependent kinase and dephosphorylation of the retinoblastoma protein, which results in early G1 phase cell cycle arrest.42, 43 A different mechanism involves altered expression of cyclin A and D, and p27/Kip1, again resulting in reduced cyclin-dependent kinase activity and cell cycle arrest.43
Apoptosis
There are two mechanisms leading to apoptosis: the death receptor pathway and the intrinsic pathway.44 Although exposure to romidepsin and other HDAC inhibitors can lead to hyper-acetylation of death receptor promoters, including tumor-necrosis factor-related apoptosis-inducing ligand, death receptor 5, Fas ligand and Fas,45 the significance of this mechanism does not seem to be universally acknowledged. Some leukemia cells do not show induction of the tumor-necrosis factor-related apoptosis-inducing ligand or Fas pathways, or undergo apoptosis after exposure to HDAC inhibitors.46 The intrinsic pathway involves the perturbation of mictochondrial membranes resulting in the generation of reactive oxygen species. In normal cells exposed to HDAC inhibitors, reactive oxygen species do not accumulate because of breakdown by thioredoxin; some tumor cells that do not express the thioredoxin gene accumulate reactive oxygen species and undergo apoptosis.44
Romidepsin has also been shown to increase acetylation of the HSP 90 chaperone protein, causing proteasomal degradation and inducing apoptosis.47 The proteasome inhibitor bortezomib appears to work synergistically with HDAC inhibitors,48 but it is interesting that the HSP 90 inhibitor, geldanamycin, antagonizes HDAC inhibitor activity.49
Angiogenesis inhibition
Romidepsin inhibits hypoxia-induced angiogenesis of endothelieal cells in vitro, but does not cause cytotoxicity. It decreased angiogenesis in a chick embryo model, with no signs of thrombosis or hemorrhage, and inhibited angiogenesis strongly in mouse model tumors.50 Romidepsin appears to reduce angiogenic-stimulating factors such as vascular endothelial growth factors, vascular endothelial growth factor receptor, FLT1 and FLK1, and increases induction of angiogenic inhibitory factors VHL and neurofibromin 2;50 however, the correlation between anti-vascular endothelial growth factor activity and in vivo efficacy has not been established.
Pharmacology
Pharmacokinetics
A two-compartment model with linear kinetics can be used in the pharmacokinetic analysis of romidepsin.51, 52 Two dosing schedules were studied in separate phase I trials. One schedule involved a 4-h infusion on days 1 and 5 of a 21-day cycle, where the maximum tolerated dose (MTD) was determined as 17.8 mg m–2,52 while the second involved a 4-h infusion on days 1, 8 and 15 of a 28-day cycle, where the MTD was determined as 13.3 mg m–2.53 With both schedules, romidepsin exhibited linear pharmacokinetics up to the MTD.52, 53 Total clearance volumes in adults have been reported as 4.8 l h–1 m–2 at a 13 mg m–2 dose,54and 10.5 l h–1 m–2 at a 17.8 mg m–2 dose.52 A phase I study in pediatric patients (2–21 years, median age 13) with refractory solid tumors who received romidepsin in 4-h infusions of 17 mg m–2 reported a clearance volume of 6.8 l h–1 m–2.55 The elimination half-life has been reported as 3.5 h51 and 3.67 h54 at 13 mg m–2, and 8.1 h52 at 17.8 mg m–2.
Pharmacodynamics
A typical assay for pharmacodynamic studies is to monitor histone acetylation of peripheral blood mononuclear cells (PBMCs).52, 55, 56 Increased acetylation is observed in PBMCs of patients after treatment with romidepsin, with maximal accumulation of acetyl H3 histones in PBMCs occurring at 4 h after the end of an infusion.55 A study in patients with acute myelogenous leukemia and chronic lymphocytic leukemia using a 4 h 13 mg m–2 infusion on days 1, 8 and 15 of a 28-day cycle reported 100% histone acetylation of H3 and H4 histones for all chronic lymphocytic leukemia patients after 4 h, and for six out of seven chronic lymphocytic leukemia patients after 24 h.54 This study also reported an increase in p21 protein expression concurrent with H4 acetylation of the p21 promoter gene.54
A phase II study of T-cell lymphoma patients monitored several biomarkers: PBMCs histone acetylation, ABCB1 gene expression in PBMCs, ABCB1 gene expression in biopsy samples and blood fetal hemoglobin (HbF) levels.57 A global increase in PBMCs histone acetylation was reported in 73% of patients within 4 h of treatment, and in 40% of patients after 24 to 48 h.57 The number of patients having a twofold or higher than baseline ABCB1 expression in PBMCs was 56% at 4 h, and 30% at 48 h.57 A fourfold or greater increase in circulating HbF was reported in 60% of the patients. The histone H3 acetylation in PBMCs at the 24-h time point appeared to correlate with response; there was no correlation between the levels of ABCB1 induction and pharmacokinetic parameters, or between ABCB1 induction in biopsy samples and clinical disease response.57 These data suggested that peak drug concentration (Cmax) and overall exposure were important in determining response.17, 57
T-cell lymphoma clinical trials
In preclinical studies, greater antitumor activity was observed with intermittent administration than with daily administration.52 Short infusions (>30 s to 4 min) and prolonged infusions (>24 h) caused greater toxicity than infusions of 1–4 h.52 Therefore, in phase I trials, romidepsin was tested using a 4-h i.v. infusion. Patients who consent to be treated in phase I trials have cancers for which no known standard therapy exists, or such therapy has already failed, so that patients are not denied any curative or definitely life-extending options. One phase I study52 administered a 4-h infusion on days 1 and 5 of a 21-day cycle; the MTD was defined at 17.8 mg m–2 with dose-limiting toxicity manifest as grade-3 fatigue, grade-3 nausea and vomiting, grade-4 thrombocytopenia (low platelet levels) and grade-4 cardiac arrhythmia. Another phase I study53 administered romidepsin in a 4-h infusion on days 1, 8 and 15 of a 28-day cycle; the MTD in this trial was defined at 13.3 mg m–2 with dose-limiting toxicity manifesting as grade 3 thrombocytopenia and fatigue. A partial response was seen in a patient with renal cell carcinoma.52 Several patients with T-cell lymphoma (cutaneous or peripheral) exhibited significant reductions in skin lesions and tumor size after treatment with romidepsin.53, 58
Owing to the dramatic responses seen in phase I trials, a phase II trial to define the response rate and toxicity profile in patients with T-cell lymphoma was initiated.40 Romidepsin was administered via a 4-h infusion on days 1, 8 and 15 of a 28-day cycle. Complete responses were observed in 4 patients (6%), and partial responses were observed in 20 patients (28%).59 The toxicities observed were consistent with those reported previously, including extreme fatigue, nausea and vomiting.52 Granulocytopenia (failure of bone marrow to make white blood cells) and thrombocytopenia (low platelet counts) were observed as well, with values returning to baseline by the next cycle of treatment.59
Clinical trials of romidepsin
Table 1 highlights the published clinical trial data on romidepsin; all of the phase I trials were on patients who had non-responsive cancers. The favorable results of the NCI-sponsored multi-institutional phase II trial,51, 57, 59, 60 followed by consistent results by the pivotal phase II trial conducted by Gloucester Pharmaceuticals61 led to the approval by the US FDA in November of 2009.
Toxicity and side effects
Cardiac toxicities were a primary concern in the early clinical trials because of preclinical results, leading to electrocardiograms (ECGs) and other measures related to cardiac function being taken before and after every treatment.52 Many of the early toxicities were abnormalities of the ECG waveform, which represent repolarization abnormalities in the electric functioning of the heart; grade 1 T-wave flattening or grade 2 ST segment depression was observed in more than half of the ECGs, but although these abnormalities can also be indicative of cardiac ischemia, in the case of romidepsin this was not so, because they were not associated with elevation of cardiac troponin or with altered left ventricular function.60 Further follow-up over 3 years with a larger body of clinical data confirms that the ECG abnormalities are not clinically significant nor indicative of any real cardiac toxicity.39 Therefore, this observation is no longer a clinical concern, except that patients are screened with an ECG to check for pre-existing cardiac rhythm abnormalities, including the very rare congenital long QT syndrome, which is a condition where the interval between the Q-wave and the T-wave on the ECG is prolonged, and this can lead to further, even fatal, problems if the electrical circuitry of the heart is further affected in these patients.
The majority of patients receiving romidepsin experience nausea, vomiting and anorexia,17, 52, 54, 59, 65 and Grant et al.17 note that the antiemetic routine followed at the NCI was to administer 1 mg granisetron i.v. before romidepsin, followed by 1 mg orally every 12 h for 3 days.17 Progressive fatigue and occasional fever were also noted with romidepsin.52, 53, 54, 59, 65 Also of concern are the hematological side effects; regular blood tests are administered to patients on romidepsin to monitor these symptoms. (This information comes from the package insert.) Leukopenia, granulocytopenia and thrombocytopenia were all observed; in each case the effect is quickly reversed after cessation of the treatment.17, 59
Synthetic studies
The main challenges in romidepsin synthesis include the asymmetric construction of the hydroxy mercapto heptenoic acid unit, the 16-membered cyclic depsipeptide ring itself (that is, four amino acids and hydroxy mercapto heptenoic acid), and the intramolecular oxidative coupling of the thiol moieties to produce the stable prodrug form of the molecule. The total synthesis was first completed in 1996 in 14 steps with an 18% overall yield.67 An improved synthesis utilizing nine steps with a 13% overall yield was published in 2007,68 using, as in the first scheme, an asymmetric acetate aldol reaction and a lactonization step for macrolization. In 2008, Wen et al.69 utilized a lactamization as an alternative route to cyclization, reporting increased cyclization efficiency. However, due both to the chemical complexity of synthesis and the low yield of the producing organism, development was hindered mainly by shortages of the product.70 Therefore, attempts have been made to develop active analogs.
Using a series of synthetic analogs, Yurek-George et al.71 were able to examine the importance of several features of the romidepsin structure. They concluded that while the thiol group at position 13 does indeed function as a Zn-binding group within the HDAC active site, the cysteine residue is non-essential, provided that the Zn-binding thiol remains protected as in the oxidized pro-drug form.71 This assertion is supported by work done with linear compounds in which the thiol was masked as a thioester, undergoing enzymatic hydrolysis in the cell to produce the free thiol.72, 73 Yurek-George et al. also found that the unsaturated Z-dehydrobutyrine residue, which could be susceptible to a Michael addition, could be replaced by less reactive side chains (for example, 3 and 4, Figure 2) with no loss in in vitro potency, but that the macrocyclic scaffold itself was an essential component of HDAC inhibition.71 However, several small methyl ester structures without the macrocyclic backbone have proved to induce hyperacetylation in Drosophila S2 cells.74 These structures retain the disulfide link of romidepsin, thus preserving its prodrug quality. The analogs studied consisted of several small cyclic disulfides; the number of methylene units in the ring was varied to provide a range of lengths for the linker from the ester to the reduced thiol group.74 Studies with HDAC inhibitors trichostatin A and suberoylanilide hydroxamic acid (SAHA) showed important interactions between the drugs and several phenylalanine residues surrounding the enzyme active site pocket,38 so the methyl ester was used to attach various lipophilic caps, in an effort to improve enzyme interaction.74

Structures of two romidepsin analogs (compounds 3 and 4), where the Z-dehydrobutyrine (Z-Dhb) residue, which may be susceptible to a Michael addition, has been replaced with a valine moiety.71
Despite these interesting medicinal chemistry studies, to the best of our knowledge, no analogs of romidepsin are in advanced stages of development.
Conclusions
Romidepsin, a potent HDAC inhibitor, offers a promising new treatment for a disease with few existing therapies. This natural product was approved by the US FDA in November of 2009 for use in patients with CTLC under the trade name Istodax marketed by Celgene Corporation. The dosing schedule was approved as 14 mg m–2 on days 1, 8 and 15 of a 28-day cycle.(This information comes from the package insert.) After a year of approval as an antineoplastic agent, preliminary information suggests that romidepsin is finding a real place in the treatment of CTCL, and the activity and manageable toxicity are helpful for both patients receiving treatment and doctors prescribing it (personal communication by several medical doctors to WM). At the time of preparation of this review, a supplemental new drug application was being prepared for peripheral T-cell lymphoma,75 and only time will tell if romidepsin's use expands to the treatment of other cancers.
References
Kurdistani, S. K. Histone modifications as markers of cancer prognosis: a cellular view. Br. J. Cancer 97, 1–5 (2007).
Emanuele, S., Lauricella, M. & Tesoriere, G. Histone deacetylase inhibitors: apoptotic effects and clinical implications (review). Int. J. Oncol. 33, 637–646 (2008).
Glaser, K. B. HDAC inhibitors: clinical update and mechanism-based potential. Biochem. Pharmacol. 74, 659–671 (2007).
Zheng, Y. G., Wu, J., Chen, Z. & Goodman, M. Chemical regulation of epigenetic modifications: opportunities for new cancer therapy. Med. Res. Rev. 28, 645–687 (2008).
Kuo, M. H. & Allis, C. D. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays 20, 615–626 (1998).
Fukuda, H., Sano, N., Muto, S. & Horikoshi, M. Simple histone acetylation plays a complex role in the regulation of gene expression. Brief Funct. Genomic. Proteomic. 5, 190–208 (2006).
Mitsiades, C. S. et al. Transcriptional signature of histone deacetylase inhibition in multiple myeloma: biological and clinical implications. Proc. Natl Acad. Sci. USA 101, 540–545 (2004).
Melnick, A. & Licht, J. D. Histone deacetylases as therapeutic targets in hematologic malignancies. Curr. Opin. Hematol. 9, 322–332 (2002).
Mahlknecht, U. & Hoelzer, D. Histone acetylation modifiers in the pathogenesis of malignant disease. Mol. Med. 6, 623–644 (2000).
Marks, P. A., Richon, V. M., Breslow, R. & Rifkind, R. A. Histone deacetylase inhibitors as new cancer drugs. Curr. Opin. Oncol. 13, 477–483 (2001).
Klisovic, M. I. et al. Depsipeptide (FR 901228) promotes histone acetylation, gene transcription, apoptosis and its activity is enhanced by DNA methyltransferase inhibitors in AML1/ETO-positive leukemic cells. Leukemia 17, 350–358 (2003).
Bieliauskas, A. V. & Pflum, M. K. Isoform-selective histone deacetylase inhibitors. Chem. Soc. Rev. 37, 1402–1413 (2008).
Papeleu, P. et al. Differential effects of histone deacetylase inhibitors in tumor and normal cells-what is the toxicological relevance? Crit. Rev. Toxicol. 35, 363–378 (2005).
Dokmanovic, M. & Marks, P. A. Prospects: histone deacetylase inhibitors. J. Cell. Biochem. 96, 293–304 (2005).
Ueda, H. et al. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum no. 968. I. Taxonomy, fermentation, isolation, physico-chemical and biological properties, and antitumor activity. J. Antibiot. 47, 301–310 (1994).
Xiao, J. J., Byrd, J., Marcucci, G., Grever, M. & Chan, K. K. Identification of thiols and glutathione conjugates of depsipeptide FK228 (FR901228), a novel histone protein deacetylase inhibitor, in the blood. Rapid Commun. Mass Spectrom. 17, 757–766 (2003).
Grant, C. et al. Romidepsin: a new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Expert Rev. Anticancer. Ther. 10, 997–1008 (2010).
Willemze, R. et al. WHO-EORTC classification for cutaneous lymphomas. Blood 105, 3768–3785 (2005).
Whittaker, S. J., Marsden, J. R., Spittle, M. & Russell Jones, R. Joint British Association of Dermatologists and UK Cutaneous Lymphoma Group guidelines for the management of primary cutaneous T-cell lymphomas. Br. J. Dermatol. 149, 1095–1107 (2003).
Litvinov, I. V., Jones, D. A., Sasseville, D. & Kupper, T. S. Transcriptional profiles predict disease outcome in patients with cutaneous T-cell lymphoma. Clin. Cancer Res. 16, 2106–2114 (2010).
Weinstock, M. A. & Horm, J. W. Mycosis fungoides in the United States: increasing incidence and descriptive epidemiology. JAMA 260, 42–46 (1988).
Criscione, V. D. & Weinstock, M. A. Incidence of cutaneous T-cell lymphoma in the United States, 1973–2002. Arch. Dermatol. 143, 854–859 (2007).
Kim, E. J. et al. Immunopathogenesis and therapy of cutaneous T cell lymphoma. J. Clin. Invest. 115, 798–812 (2005).
Lorincz, A. L. Cutaneous T-cell lymphoma (mycosis fungoides). Lancet 347, 871–876 (1996).
Kim, Y. H., Liu, H. L., Mraz-Gernhard, S., Varghese, A. & Hoppe, R. T. Long-term outcome of 525 patients with mycosis fungoides and Sézary syndrome: clinical prognostic factors and risk for disease progression. Arch. Dermatol. 139, 857–866 (2003).
Bradford, P. T., Devesa, S. S., Anderson, W. F. & Toro, J. R. Cutaneous lymphoma incidence patterns in the United States: a population-based study of 3884 cases. Blood 113, 5064–5073 (2009).
Kim, Y. H., Chow, S., Varghese, A. & Hoppe, R. T. Clinical characteristics and long-term outcome of patients with generalized patch and/or plaque (T2) mycosis fungoides. Arch. Dermatol. 135, 26–32 (1999).
Duvic, M. et al. Analysis of long-term outcomes of combined modality therapy for cutaneous T-cell lymphoma. J. Am. Acad. Dermatol. 49, 35–49 (2003).
Zackheim, H. S., Amin, S., Kashani-Sabet, M. & McMillan, A. Prognosis in cutaneous T-cell lymphoma by skin stage: long-term survival in 489 patients. J. Am. Acad. Dermatol. 40, 418–425 (1999).
Siegel, R. S., Pandolfino, T., Guitart, J., Rosen, S. & Kuzel, T. M. Primary cutaneous T-cell lymphoma: review and current concepts. J. Clin. Oncol. 18, 2908–2925 (2000).
Shigematsu, N. et al. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. II. Structure determination. J. Antibiot. 47, 311–314 (1994).
Manoharan, T. H., Burgess, J. A., Ho, D., Newell, C. L. & Fahl, W. E. Integration of a mutant c-Ha-ras oncogene into C3H/10T1/2 cells and its relationship to tumorigenic transformation. Carcinogenesis 6, 1295–1301 (1985).
Wang, R., Brunner, T., Zhang, L. & Shi, Y. Fungal metabolite FR901228 inhibits c-Myc and Fas ligand expression. Oncogene 17, 1503–1508 (1998).
Rajgolikar, G., Chan, K. K. & Wang, H. C. Effects of a novel antitumor depsipeptide, FR901228, on human breast cancer cells. Breast Cancer Res. Treat. 51, 29–38 (1998).
Nakajima, H., Kim, Y. B., Terano, H., Yoshida, M. & Horinouchi, S. FR901228, a potent antitumor antibiotic, is a novel histone deacetylase inhibitor. Exp. Cell Res. 241, 126–133 (1998).
Yoshida, M. et al. Histone deacetylase as a new target for cancer chemotherapy. Cancer Chemother. Pharmacol. 48 (Suppl 1), S20–S26 (2001).
Furumai, R. et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 62, 4916–4921 (2002).
Finnin, M. S. et al. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 401, 188–193 (1999).
Piekarz, R. & Bates, S. A review of depsipeptide and other histone deacetylase inhibitors in clinical trials. Curr. Pharm. Des. 10, 2289–2298 (2004).
Archer, S. Y., Meng, S., Shei, A. & Hodin, R. A. p21WAF1 is required for butyrate-mediated growth inhibition of human colon cancer cells. Proc. Natl Acad. Sci. USA 95, 6791–6796 (1998).
Burgess, A. J. et al. Up-regulation of p21WAF1/CIP1 by histone deacetylase inhibitors reduces their cytotoxicity. Mol. Pharmacol. 60, 828–837 (2001).
Rosato, R. R., Almenara, J. A. & Grant, S. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res. 63, 3637–3645 (2003).
Sandor, V. et al. p21-dependent G1 arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br. J. Cancer 83, 817–825 (2000).
Peart, M. J. et al. Novel mechanisms of apoptosis induced by histone deacetylase inhibitors. Cancer Res. 63, 4460–4471 (2003).
Insinga, A. et al. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat. Med. 11, 71–76 (2005).
Nebbioso, A. et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat. Med. 11, 77–84 (2005).
Yu, X. et al. Modulation of p53, ErbB1, ErbB2, and Raf-1 expression in lung cancer cells by depsipeptide FR901228. J. Natl Cancer Inst. 94, 504–513 (2002).
Yu, C. et al. The proteasome inhibitor bortezomib interacts synergistically with histone deacetylase inhibitors to induce apoptosis in Bcr/Abl+ cells sensitive and resistant to STI571. Blood 102, 3765–3774 (2003).
Huang, H. C., Liu, Y. C., Liu, S. H., Tzang, B. S. & Lee, W. C. Geldanamycin inhibits trichostatin A-induced cell death and histone H4 hyperacetylation in COS-7 cells. Life Sci. 70, 1763–1775 (2002).
Kwon, H. J., Kim, M. S., Kim, M. J., Nakajima, H. & Kim, K. W. Histone deacetylase inhibitor FK228 inhibits tumor angiogenesis. Int. J. Cancer 97, 290–296 (2002).
Woo, S. et al. Population pharmacokinetics of romidepsin in patients with cutaneous T-cell lymphoma and relapsed peripheral T-cell lymphoma. Clin. Cancer Res. 15, 1496–1503 (2009).
Sandor, V. et al. Phase I trial of the histone deacetylase inhibitor, depsipeptide (FR901228, NSC 630176), in patients with refractory neoplasms. Clin. Cancer Res. 8, 718–728 (2002).
Marshall, J. L. et al. A phase I trial of depsipeptide (FR901228) in patients with advanced cancer. J. Exp. Ther. Oncol. 2, 325–332 (2002).
Byrd, J. C. et al. A phase 1 and pharmacodynamic study of depsipeptide (FK228) in chronic lymphocytic leukemia and acute myeloid leukemia. Blood 105, 959–967 (2005).
Fouladi, M. et al. Phase I study of depsipeptide in pediatric patients with refractory solid tumors: a Children's Oncology Group report. J. Clin. Oncol. 24, 3678–3685 (2006).
Klimek, V. M. et al. Tolerability, pharmacodynamics, and pharmacokinetics studies of depsipeptide (romidepsin) in patients with acute myelogenous leukemia or advanced myelodysplastic syndromes. Clin. Cancer Res. 14, 826–832 (2008).
Bates, S. E. et al. Laboratory correlates for a phase II trial of romidepsin in cutaneous and peripheral T-cell lymphoma. Br. J. Haematol. 148, 256–267 (2009).
Piekarz, R. L. et al. Inhibitor of histone deacetylation, depsipeptide (FR901228), in the treatment of peripheral and cutaneous T-cell lymphoma: a case report. Blood 98, 2865–2868 (2001).
Piekarz, R. L. et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J. Clin. Oncol. 27, 5410–5417 (2009).
Piekarz, R. L. et al. Cardiac studies in patients treated with depsipeptide, FK228, in a phase II trial for T-cell lymphoma. Clin. Cancer Res. 12, 3762–3773 (2006).
Whittaker, S. J. et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 28, 4485–4491 (2010).
Odenike, O. M. et al. Histone deacetylase inhibitor romidepsin has differential activity in core binding factor acute myeloid leukemia. Clin. Cancer Res. 14, 7095–7101 (2008).
Whitehead, R. P. et al. Phase II trial of romidepsin (NSC-630176) in previously treated colorectal cancer patients with advanced disease: a Southwest Oncology Group study (S0336). Invest. New Drugs 27, 469–475 (2009).
Schrump, D. S. et al. Clinical and molecular responses in lung cancer patients receiving romidepsin. Clin. Cancer Res. 14, 188–198 (2008).
Stadler, W. M., Margolin, K., Ferber, S., McCulloch, W. & Thompson, J. A. A phase II study of depsipeptide in refractory metastatic renal cell cancer. Clin. Genitourin. Cancer 5, 57–60 (2006).
Molife, L. R. et al. Phase II, two-stage, single-arm trial of the histone deacetylase inhibitor (HDACi) romidepsin in metastatic castration-resistant prostate cancer (CRPC). Ann. Oncol. 21, 109–113 (2010).
Li, K. W., Wu, J., Xing, W. N. & Simon, J. A. Total synthesis of the antitumor depsipeptide FR-901,228. J. Am. Chem. Soc. 118, 7237–7238 (1996).
Greshock, T. J., Johns, D. M., Noguchi, Y. & Williams, R. M. Improved total synthesis of the potent HDAC inhibitor FK228 (FR-901228). Org. Lett. 10, 613–616 (2008).
Wen, S., Packham, G. & Ganesan, A. Macrolactamization versus macrolactonization: total synthesis of FK228, the depsipeptide histone deacetylase inhibitor. J. Org. Chem. 73, 9353–9361 (2008).
Jones, P. & Steinkuhler, C. From natural products to small molecule ketone histone deacetylase inhibitors: development of new class specific agents. Curr. Pharm. Des. 14, 545–561 (2008).
Yurek-George, A. et al. The first biologically active synthetic analogues of FK228, the depsipeptide histone deacetylase inhibitor. J. Med. Chem. 50, 5720–5726 (2007).
Suzuki, T. et al. Novel inhibitors of human histone deacetylases: design, synthesis, enzyme inhibition, and cancer cell growth inhibition of SAHA-based non-hydroxamates. J. Med. Chem. 48, 1019–1032 (2005).
Suzuki, T. et al. Identification of a potent and stable antiproliferative agent by the prodrug formation of a thiolate histone deacetylase inhibitor. Bioorg. Med. Chem. Lett. 17, 1558–1561 (2007).
Mays, J. R., Restituyo, J. A., Katzenberger, R. J., Wassarman, D. A. & Rajski, S. R. Cyclic disulfides as functional mimics of the histone deacetylase inhibitor FK-228. Tetrahedron Lett. 48, 4579–4583 (2007).
Pro, B. et al. Final results from a pivotal, multicenter, international, open-label, phase 2 study of romidepsin in progressive or relapsed peripheral T-cell lymphoma (PTCL) following prior systemic therapy. 52nd American Society of Hematology Annual Meeting and Exposition Abstract 114 Orange County Convention Center, Orlando, FL, USA (2010).
Acknowledgements
This review was prepared in support of grant P01-CA125066 from the National Cancer Institute/National Institutes of Health, Bethesda, MD, USA. Some details were added (by WM) with permission from Celgene Corporation. We thank Drs Sloan Ayers, Mario Figueroa, James McAlpine and Arlene Sy-Cordero for helpful comments.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
VanderMolen, K., McCulloch, W., Pearce, C. et al. Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): a natural product recently approved for cutaneous T-cell lymphoma. J Antibiot 64, 525–531 (2011). https://doi.org/10.1038/ja.2011.35
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2011.35
Keywords
This article is cited by
-
Chemoenzymatic tandem cyclization for the facile synthesis of bicyclic peptides
Communications Chemistry (2024)
-
Progress in discovery and development of natural inhibitors of histone deacetylases (HDACs) as anti-cancer agents
Naunyn-Schmiedeberg's Archives of Pharmacology (2024)
-
Establishment and characterization of NCC-DFSP5-C1: a novel patient-derived dermatofibrosarcoma protuberans cell line
Human Cell (2024)
-
Romidepsin-induced durable remission for relapsed nodal peripheral T-cell lymphoma with T follicular helper phenotype after allogeneic hematopoietic cell transplantation
International Journal of Hematology (2023)
-
Establishment and characterization of NCC-PMP2-C1: a novel patient-derived cell line of pseudomyxoma peritonei with signet ring cells
Human Cell (2023)
