
Abstract
Four new alkaloids, including two new meleagrin analogs, meleagrin D (1) and E (2), and two new diketopiperazines, roquefortine H (3) and I (4), were isolated from a deep ocean sediment-derived fungus Penicillium sp. Meleagrin D (1) and E (2) possess unprecedented acetate–mevalonate-derived side chains on the imidazole moiety. These new meleagrins showed weak cytotoxicity against the A-549 cell line, whereas meleagrin B (5) and meleagrin (6), which were isolated previously from the same strain, induced HL-60 cell apoptosis or arrested the cell cycle through G2/M phase, respectively. The results indicate that the distinct substitutions on the imidazole ring significantly influence the cytotoxicity of the meleagrin alkaloids.
Similar content being viewed by others

Introduction
The meleagrin and roquefortine groups, mainly isolated from Penicillium species, are biogenetically interrelated alkaloids1, 2, 3, 4, 5, 6, 7 with promising biological properties, such as neurotoxic,1 antibacterial,8 cytochrome P450 inhibitory9 and tubulin polymerization inhibitory activity.10 We have previously reported on meleagrin B (5), a novel complex compound composed of a meleagrin alkaloid moiety and a rare diterpene moiety, from a deep ocean sediment-derived fungus Penicillium sp. F23-2.11 Meleagrin B (5) showed potent cytotoxicity against four tumor cell lines, with IC50 values ranging from 1.8 to 6.7 μM. In an effort to obtain more insight into the biosynthetic mechanisms and structure–activity relationships in this family of metabolites, we investigated the active constituents of this fungus, which led to the isolation of four new alkaloids, meleagrin D (1), meleagrin E (2), roquefortine H (3) and roquefortine I (4). Compounds 1 and 2 possess unprecedented side chains, which originated from six acetates via the acetate-mevalonate pathway, on the imidazole moiety. They showed weak cytotoxicity against the A-549 cell line, while the alkaloid nucleus, meleagrin (6), exhibited moderate cytotoxicity against the A-549 and HL-60 cell lines, with the IC50 values being 19.9 and 7.4 μM, respectively. Comparison with the previous antitumor evaluation of meleagrin B (5) and C11 suggests that addition of the acetate–mevalonate-derived C5 or C9 side chains on N-17 negatively affects the activities of meleagrin alkaloids, while the diterpene substitution on the imidazole ring enhances the cytotoxic activities. On further exploring the cytotoxic mechanisms of meleagrin B (5) and meleagrin (6) by flow cytometric analysis, meleagrin B (5) induced the HL-60 cell apoptosis at 5 and 10 μM, while meleagrin (6) arrested the cell cycle through G2/M phase at the same concentrations. This analysis indicates that addition of the diterpene moiety on the imidazole ring endows the molecules with distinct cytotoxic mechanisms. In this paper, we describe the isolation, structure elucidation and analysis of the cytotoxicity against A-549 and HL-60 cell lines of the new compounds, and the flow cytometric analysis of meleagrin B (5) and meleagrin (6) on the HL-60 cell line.
Results and discussion
Structure determination
Compound 1 was obtained as a yellow solid. Its molecular formula (C32H38N5O5) was determined by HR-ESI-MS (m/z 572.2852 [M+H]+, calcd for 572.2873), indicating 15 degrees of unsaturation. The 1H NMR spectrum showed a characteristic ABCD system due to the aromatic resonances (δ 7.52 (1H, d, J=7.8 Hz, H-4), 7.03 (1H, t, J=7.8 Hz, H-5), 7.25 (1H, t, J=7.8 Hz, H-6), 6.95 (1H, d, J=7.8 Hz, H-7)) with the typical multiplets due to ortho- and meta-couplings. Analysis of the 13C NMR and DEPT spectra revealed 13 quaternary carbons, 10 methines, 2 methylenes and 7 methyls. Comparison of the 1H and 13C NMR data (Table 1) with those of the known compounds, meleagrin B (5),11 meleagrin (6)12 and meleagrin C,11 indicated that the structure of 1 included a N-methoxyindoline moiety (δC 65.3 (OCH3-1), 100.8 (C-2), 52.1 (C-3), 125.6 (C-3a), 124.9 (C-4), 123.6 (C-5), 128.5 (C-6), 112.1 (C-7), 146.3 (C-7a)), a 1,1-dimethyl-2-propenyl group (δH: geminal methyl, 1.23 (3H, s, CH3-24) and 1.35 (3H, s, CH3-25); terminal olefin, 6.08 (1H, brs, H-22), 5.14 (1H, brd, J=17.6 Hz, H-23a) and 5.08 (1H, brs, H-23b)/δC: 42.8 (C-21), 143.3 (C-22), 114.1 (C-23), 23.0 (C-24), 21.7 (C-25)), a histidine residue (δH: 8.45 (1H, s, H-15), 7.62 (1H, s, H-18), 8.59 (1H, s, H-20)/δC: 123.6 (C-12), 163.7 (C-13), 117.7 (C-15), 135.3 (C-16), 134.4 (C-18), 122.2 (C-20)) and an α,β-unsaturated amide moiety (δH: 5.45 (1H, s, H-8)/δC: 107.1 (C-8), 141.9 (C-9), 158.8 (C-10)), which constituted a meleagrin alkaloid moiety (Figure 2). The 1H and 13C NMR spectra of 1 (Table 1) and meleagrin C11 were almost identical except for some tiny distinctions that are ascribed to the different substituents on N-19. Analysis of the HMBC correlations from CH3-6′ to C-1′, C-2′ and C-7′, from H-2′ to C-1′, C-3′, CH3-6′ and CH3-7′, from H-4′ to C-3′, C-5′, C-8′ and C-9′, and from CH3-8′ to C-4′, C-5′ and C-9′ could afford an unprecedented 2,6-dimethylhept-2-en-4-one moiety connecting with the imidazole ring through the C-5′–N-19 single bond, which was confirmed by the NOESY correlations between H-20 and H-8′, H-9′, and 2H-4′ (Figure 2). Thus the planar structure of 1 was established, namely meleagrin D (Figure 1). The absolute configuration of meleagrin (6), isolated from the same species, has been well established.11, 12 By comparison of the 1D NMR data and the optical rotation values of meleagrin D (1) and meleagrin (6) with those reported for meleagrin,11, 12 the absolute configurations of C-2, C-3 and the C-12/C-15 double bond in 1 were deduced to be consistent with those in 6 on biogenetic grounds.

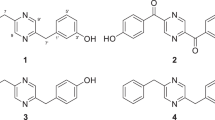
Structures of compounds 1–6.
Compound 2 was a yellow solid with the molecular formula C32H39N5O6, established by HR-ESI-MS (m/z 590.2980 [M+H]+, calcd for 590.2979). A careful comparison of the 1H and 13C NMR data with those of compound 1 (Table 1) indicated that they had the same meleagrin moiety, but with the structure of the N-19 substituent changed. The 1D NMR signals of the sp2 quaternary carbon (δ 157.1) and the sp2 methine (δH 5.78 (1H, s)/δC 124.1) were replaced by those of an oxygen-bearing C-1′ (δ 69.7) and a methylene CH2-2′ (δH 2.40 (1H, d, J=16.1 Hz), 2.35 (1H, d, J=16.1 Hz)/δC 54.9), and the chemical shifts of C-3′, CH3-6′ and CH3-7′ changed significantly (Table 1). It indicated that a novel 2-hydroxy-2,6-dimethylheptan-4-one moiety, also confirmed by the HMBC correlations (Figure 2), was attached to the imidazole ring through the C-5′–N-19 single bond. The structure of 2 was established as a new meleagrin analog, namely meleagrin E (Figure 1).

Key 1H–1H COSY, HMBC and NOESY correlations of compounds 1–4.
Compound 3 was a yellow solid. Its molecular formula was established as C28H33N5O3 by HR-ESI-MS (m/z 488.1674 [M+H]+, calcd for 488.2662). The 1H NMR spectrum showed a characteristic ABCD system due to the aromatic resonances (δ 6.60 (1H, d, J=7.7 Hz, H-7), 7.10 (1H, t, J=7.7 Hz, H-8), 6.76 (1H, t, J=7.7 Hz, H-9), 7.17 (1H, d, J=7.7 Hz, H-10)) and an ABX system assigned to the protons that comprised the exocyclic double bond (δ 5.99 (1H, dd, J=10.1, 17.3 Hz, H-19), 5.13 (1H, d, J=10.1 Hz, H-20a), 5.10 (1H, d, J=17.3 Hz, H-20b)). Analysis of the 13C NMR and DEPT spectra revealed 10 quaternary carbons, 10 methines, 3 methylenes and 5 methyls. The 1H–1H COSY and HMBC spectra (Figure 2) confirmed that compound 3 has the same planar structure as roquefortine C,1 the biogenetic precursor of meleagrin, except for a minor substituent variation on N-16. The structure of the substitute was established as 4-methylpentan-2-one by examining the HMBC correlations from CH3-1′ to C-2′ and C-3′; from H-3′ to C-2′, C-4′, C-5′ and C-6′; and from CH3-5′ to C-3′, C-4′ and C-6′. Comparison of the 1D NMR spectra (Table 2) with those of meleagrin C11 further confirmed the same substituent system on the imidazole ring. Thus the planar structure of 3 was established, namely roquefortine H (Figure 1), and its absolute configurations of C-5a, C-10b, C-11a and the C-3/C-12 double bond, by comparison of the 1D NMR data and the optical rotation values, were proposed to be the same as roquefortine C, the absolute configuration of which has been established.13
Compound 4, a yellow solid, had the molecular formula C23H25N5O3, as established by HR-ESI-MS (m/z 420.2030 [M+H]+, calcd for 420.2036). Comparison of its 1D NMR spectra with those of roquefortine C1 and compound 3 indicated that they had the same skeleton and that N-16 in 4 was not substituted, but the H-11a (δ 4.05 in 3) was replaced by an OCH3 (δH 2.84, δC 52.0). This structure was supported by the HMBC correlation from OCH3-11a to C-11a (δ 91.2) and the doublet peaks of H-11α and H-11β. On further comparing the 1D NMR spectra with those of roquefortine G,11 they were found to have the same OCH3-11a substitute and the chemical shifts for H-11α, H-11β, C-11, C-11a, C-1 and OCH3-11a (Table 2)11 were identical. As the absolute configuration of C-11a in roquefortine G has been established,11 the absolute configuration of compound 4 was deduced to be the same as that of roquefortine G on biogenetic ground and by their similar 1H and 13C shifts. The structure of 4 was established as a new roquefortine C analog, namely roquefortine I (Figure 1).
The biogenetic relationship of all the new alkaloids was postulated in a plausible route (Scheme 1). Biosynthetically, the unprecedented side chains of compounds 1 and 2 are proposed to originate from six acetates via the acetate–mevalonate pathway. Intermolecular Michael addition11 of intermediates (b) or (c) toh meleagrin (6), of which the biosynthetic pathway has been well established,14, 15, 16 would provide meleagrin E (2) and D (1), respectively.

Postulated biosynthetic pathway of compounds 1–4.
Cytotoxic activities
The new compounds 1–4 and meleagrin (6) were evaluated for their cytotoxicity against the HL-60 cell line by the MTT method17 and against the A-549 cell line by the SRB method.18 Meleagrin D (1) and E (2) showed weak cytotoxicity against the A-549 cell line, while meleagrin (6) exhibited moderate cytotoxicity against the A-549 and HL-60 cell lines, with the IC50 values being 19.9 and 7.4 μM, respectively (Table 3). Comparison with the previous antitumor evaluation of meleagrin B (5) and C11 indicated that addition of the acetate–mevalonate-derived C5 or C9 side chains on N-17 negatively affects the activities of meleagrin alkaloids, while the diterpene substitution on the imidazole ring enhances the cytotoxic activities. The 9-O-methyl analog of meleagrin, oxaline, was reported to inhibit tubulin polymerization, resulting in cell cycle arrest through the M phase in Jurkat cells.10 Therefore, we further explored the potential cytotoxic mechanisms of meleagrin B (5) and meleagrin (6) by flow cytometric analysis.19 Meleagrin B (5) induced HL-60 cell apoptosis at 5 and 10 μM, while meleagrin (6) arrested the cell cycle through G2/M phase at the same concentrations (Figure 3). The results indicted that meleagrin (6), similar to oxaline, is also an inhibitor of tubulin polymerization. Interestingly, addition of the diterpene moiety on the imidazole ring produces distinct cytotoxic mechanisms. It warrants further investigation on the real molecular targets of the novel alkaloid–diterpene complex compound meleagrin B (5).

Flow cytometric analysis of compounds 5 and 6. HL-60 cells incubated with 5 and 6 (5, 10 μM) for 24 h were fixed and stained with PI. The percentage of cells with the hypodiploid DNA content was determined by flow cytometry. Results are representative of three separate experiments. VP16 (10 μM) was used as a positive control.
Methods
General
Specific rotations were obtained on a JASCO P-1020 digital polarimeter (JASCO Inc., Tokyo, Japan). UV spectra were recorded on Beckman DU 640 spectrophotometer (Beckman Coulter Inc., Brea, CA, USA). IR spectra were obtained on a NICOLET NEXUS 470 spectrophotometer (Thermo Electron Corporation, Madison, WI, USA) in KBr discs. 1H, 13C NMR and DEPT spectra and 2D-NMR were recorded on a JEOL JNM-ECP 600 spectrometer (JEOL Ltd., Tokyo, Japan) using TMS as internal standard, and chemical shifts were recorded as δ values. ESI-MS was measured on a Q-TOF ULTIMA GLOBAL GAA076 LC mass spectrometer (Waters Corporation, Milford, MA, USA). Semipreparative HPLC was performed using an ODS column (YMC-pak ODS-A, 10 × 250 mm, 5 μm (YMC Co. Ltd., Kyoto, Japan), 4 ml min−1).
Fungal material
The fungus, strain F23-2, was obtained from a deep ocean sediment sample (depth 5080 m). It was identified as Penicillium sp. by Professor Fengping Wang, School of Life Science & Biotechnology, Shanghai Jiao Tong University, Shanghai, China, on the basis of its ribosomal internal transcribed spacers and the 5.8S ribosomal RNA gene (ITS1-5.8S-ITS2), which was deposited in Genbank (EU770318). Working stocks were prepared on potato dextrose agar slants stored at 4 °C.
Fermentation, extraction and isolation
Spores growing on potato dextrose agar slant were inoculated into 1000-ml Erlenmeyer flasks containing 200 ml sea-water-based culture medium (potato 200 g, glucose 20 g, mannitol 20 g, maltose 10 g, peptone 5 g, yeast extract 3 g, dissolved in 1-l seawater, pH 6.0) and cultured at 28 °C for 45 days under static conditions. In all, 100 l of the whole broth gave a crude ethyl acetate extract (45.0 g), which was subjected to silica gel column chromatography (petroleum ether–acetone, v/v, gradient). The active fractions 9 and 13 eluted with the solvent of petroleum ether–acetone (6:4 and 5:5) were separately subjected to repeated Sephadex LH-20 column chromatography (chloroform–methanol, 1:1; GE Healthcare, Uppsala, Sweden). The active subfractions 13-1-1, 13-1-2, 9-7-2 and 9-10-3 were further purified, respectively, by HPLC using a reversed-phase C18 column (65/35 MeOH/H2O, 65/35 MeOH/H2O, 70/30 MeOH/H2O and 65/35 MeOH/H2O, 4.0 ml min−1), to give compounds 1 (3.2 mg), 2 (4.5 mg), 3 (3.2 mg) and 4 (5.0 mg).
Physico-chemical properties
Meleagrin D (1): yellow solid (methanol), [a]20D −116 (c. 0.01, MeOH), UV λmax (MeOH) nm (log ɛ): 200 (4.67), 231 (4.31), 341 (4.14), IR (KBr) cm−1: 2972, 2939, 1699, 1646, 1348, 1109, 976, 1H-NMR (CDCl3, 600 MHz) and 13C-NMR (CDCl3, 150 MHz), see Table 1, HR-ESI-MS m/z: 572.2852 [M+H]+ (calcd for C32H38N5O5: 572.2873).
Meleagrin E (2): yellow solid (methanol), [a]20D −55 (c. 0.01, MeOH), UV λmax (MeOH) nm (log ɛ): 200 (4.59), 221 (4.23), 340 (4.15), IR (KBr) cm−1: 2972, 2932, 1712, 1646, 1540, 1447, 1348, 1215, 1116, 917, 1H-NMR (CDCl3, 600 MHz) and 13C-NMR (CDCl3, 150 MHz), see Table 1, HR-ESI-MS m/z: 590.2980 [M+H]+ (calcd for C32H40N5O6: 590.2979).
Roquefortine H (3): yellow solid (methanol), [a]20D −430 (c. 0.01, MeOH), UV λmax (MeOH) nm (log ɛ): 200 (3.98), 218 (3.65), 314 (3.66), IR (KBr) cm−1: 2965, 2919, 1672, 1407, 1308, 1195, 1056, 1016, 917, 1H-NMR (CDCl3, 600 MHz) and 13C-NMR (CDCl3, 150 MHz), see Table 2, HR-ESI-MS m/z: 488.2674 [M+H]+ (calcd for C28H34N5O3: 488.2662).
Roquefortine I (4): yellow solid (methanol), [a]20D −285 (c. 0.01, MeOH), UV λmax (MeOH) nm (log ɛ): 200 (3.38), 226 (2.82), 326 (2.78), IR (KBr) cm−1: 2965, 2912, 1686, 1387, 1241, 1076, 917, 1H-NMR (CDCl3, 600 MHz) and 13C-NMR (CDCl3, 150 MHz), see Table 2, HR-ESI-MS m/z: 420.2030 [M+H]+ (calcd for C23H26N5O3: 420.2036).
In vitro cytotoxicity assays
The human promyelocytic leukemia HL-60 and lung adenocarcinoma A-549 cell lines were purchased from ATCC (Manassas, VA, USA). They were maintained in RPMI-1640 (HL-60) and Ham's F12K (A549) medium (GIBCO, Grand Island, NE, USA) supplemented with 10% heat-inactivated fetal bovine serum (GIBCO), L-glutamine (2 mM), penicillin (100 IU ml−1) and streptomycin (100 μg ml−1), pH 7.4 in a humidified atmosphere of 95% air plus 5% CO2 at 37 °C.
In the MTT assay, the cell line was grown in RPMI-1640 supplemented with 10% FBS under a humidified atmosphere of 5% CO2 and 95% air at 37 °C. Cell suspensions (200 μl) at a density of 5 × 104 cells ml−1 were plated in 96-well microtiter plates and incubated for 24 h. The test compound solutions (2 μl in MeOH) at different concentrations were added to each well and further incubated for 72 h under the same conditions. The MTT solution (20 μl of a 5 mg ml−1 solution in IPMI-1640 medium) was added to each well and incubated for 4 h. An old medium (150 μl) containing MTT was then gently replaced by DMSO and pipetted to dissolve any formazan crystals formed. Absorbance was then determined on a SPECTRA MAX PLUS plate reader (Molecular Devices, Sunnyvale, CA, USA) at 540 nm.
In the SRB assay, cell suspensions (200 μl) were plated in 96-cell plates at a density of 2 × 105 cells ml−1. Then the test compound solutions (2 μl in MeOH) at different concentrations were added to each well and further incubated for 24 h. Following drug exposure, the cells were fixed with 12% trichloroacetic acid and the cell layer was stained with 0.4% SRB. The absorbance of SRB solution was measured at 515 nm. Dose–response curves were generated and the IC50 values were calculated from the linear portion of log dose–response curves.
In the flow cytometric analysis, cells were plated in six-well cell culture plates (1 × 106 cells per well), in triplicate, and then treated with MeOH (control), VP16 (10 μM, positive control, purchased from Sigma (St Louis, MO, USA)) or the compounds 5 and 6 for 24 h at concentrations of 5 and 10 μM. Cells were harvested, washed twice with PBS, and the cellular DNA stained with 200 μl propidium iodide (50 μg ml−1, RNase 1 μg ml−1, Triton X-100 0.1%). After incubation at 4 °C for 20 min, the cells were analyzed using flow cytometry (Becton-Dickinson, Vantage, San Diego, CA, USA).
References
Scott, P. M., Merrien, M. A. & Polonsky, J. Roquefortine and iso-fumigaclavine A, metabolites from Penicillium roqueforti. Experientia. 32, 140–142 (1976).
Ohmomo, S., Oguma, K., Ohashi, T. & Abe, M. Isolation of a new indole alkaloid, roquefortine D, from the cultures of Penicillium roqueforti. Agric. Biol. Chem. 42, 2387–2389 (1978).
Musuku, A et al. Isolation and structure determination of a new roquefortine-related mycotoxin from Penicillium verrucosum VAR. Cyclopium isolated from Cassava. J. Nat. Prod. 57, 983–987 (1994).
Kozlovsky, A. G., Vinokurova, N. G., Solov’eva, T. F. & Buzilova, I. G. Nitrogen-containing secondary metabolites of microscopic fungi. Appl. Biochem. Microbiol. 32, 39–48 (1996).
Kozlovsky, A. G. et al. A new 16n-carboxyethyl derivative of 3,12-dihydroroquefortin. Heterocycles 60, 1639–1644 (2003).
Steyn, P. S. & Vleggaa, R. Roquefortine, an intermediate in the biosynthesis of oxaline in cultures of Penicillium oxalicum. J. Chem. Soc. Chem. Commun. 560–561 (1983).
Clark, B., Capon, R. J., Lacey, E., Tennant, S. & Gill, J. H. Roquefortine E, a diketopiperazine from an Australian isolate of Gymnoascusreessii. J. Nat. Prod. 68, 1661–1664 (2005).
Kopp, B. & Rehm, H. J. Antimicrobial action of roquefortine. Eur. J. Appl. Microbiol. Biotechnol. 6, 397–401 (1979).
Aninat, C., Hayashi, Y., Andre, F. & Delaforge, M. Molecular requirements for inhibition of cytochrome P450 activities by roquefortine. Chem. Res. Toxicol. 14, 1259–1265 (2001).
Koizumi, Y., Arai, M., Tomoda, H. & Omura, S. Oxaline, a fungal alkaloid, arrests the cell cycle in M phaseby inhibition of tubulin polymerization. Biochim. Biophys. Acta 1693, 47–55 (2004).
Du, L. et al. New alkaloids and diterpenes from a deep ocean sediment derived fungus Penicillium sp. Tetrahedron 65, 1033–1039 (2009).
Kawai, K., Nozawa, K., Nakajima, S. & Iitaka, Y. Studies on fungal products. VII. The structures of meleagrin and 9-O-p-bromobenzoylmeleagrin. Chem. Pharm. Bull. 32, 94–98 (1984).
Yamaguchi, T., Nozawa, K., Nakajima, S., Kawai, K. & Udagawa, S. Absolute configuration of roquefortine C, a tremorgenic mycotoxin. Maikotokishin 34, 29–32 (1991).
Kusch, J. & Rehm, H. J. Regulation aspects of roquefortine production by free and Ca-alginate immobilized mycelia of Penicillium roqueforti. Appl. Microbiol. Biotechnol. 23, 394–399 (1986).
Reshetilova, T. A., Vinokurova, N. G., Khmelenina, V. N. & Kozlovskii, A. G. The role of roquefortine in the synthesis of alkaloids meleagrine, glandicolines-a and glandicolines-b, and oxaline in fungi Penicillium glandicola and P.atramentosum. Microbiology 64, 27–29 (1995).
Williams, R. M., Stoching, E. M. & Sanz-Cervera, J. F. Biosynthesis of prenylated alkaloids derived from tryptophan. Top. Curr. Chem. 209, 97–173 (2000).
Mosmann, T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65, 55–63 (1983).
Skehan, P et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl Cancer Inst. 82, 1107–1112 (1990).
Du, L et al. Cytotoxic sorbicillinoids and bisorbicillinoids from a marine-derived fungus Trichoderma sp. Chem. Pharm. Bull. 57, 220–223 (2009).
Acknowledgements
This work was funded by the Chinese Ocean Mineral Resource R & D Association (DY105-2-04).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Du, L., Feng, T., Zhao, B. et al. Alkaloids from a deep ocean sediment-derived fungus Penicillium sp. and their antitumor activities. J Antibiot 63, 165–170 (2010). https://doi.org/10.1038/ja.2010.11
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2010.11
Keywords
This article is cited by
-
Marine Natural Products — a Vital Source of Novel Biotherapeutics
Current Pharmacology Reports (2022)
-
Chemical diversity and biological function of indolediketopiperazines from marine-derived fungi
Marine Life Science & Technology (2020)
-
Isolation and Antibiotic Screening of Fungi from a Hydrothermal Vent Site and Characterization of Secondary Metabolites from a Penicillium Isolate
Marine Biotechnology (2017)
-
Inhibition of biofilm in Bacillus amyloliquefaciens Q-426 by diketopiperazines
World Journal of Microbiology and Biotechnology (2016)
-
Isolation and characterization of bioactive fungi from shark Carcharodon carcharias’ gill with biopharmaceutical prospects
Chinese Journal of Oceanology and Limnology (2016)
