
Abstract
Methylated amines (MAs) are ubiquitous in the marine environment and their subsequent flux into the atmosphere can result in the formation of aerosols and ultimately cloud condensation nuclei. Therefore, these compounds have a potentially important role in climate regulation. Using Ruegeria pomeroyi as a model, we identified the genes encoding dimethylamine (DMA) monooxygenase (dmmABC) and demonstrate that this enzyme degrades DMA to monomethylamine (MMA). Although only dmmABC are required for enzyme activity in recombinant Escherichia coli, we found that an additional gene, dmmD, was required for the growth of R. pomeroyi on MAs. The dmmDABC genes are absent from the genomes of multiple marine bacteria, including all representatives of the cosmopolitan SAR11 clade. Consequently, the abundance of dmmDABC in marine metagenomes was substantially lower than the genes required for other metabolic steps of the MA degradation pathway. Thus, there is a genetic and potential metabolic bottleneck in the marine MA degradation pathway. Our data provide an explanation for the observation that DMA-derived secondary organic aerosols (SOAs) are among the most abundant SOAs detected in fine marine particles over the North and Tropical Atlantic Ocean.
Similar content being viewed by others

Introduction
Methylated amines (MAs) form part of the marine dissolved organic nitrogen pool and are ubiquitous in the marine environment. Their precursors, trimethylamine N-oxide (TMAO), glycine betaine, choline and carnitine are either osmolytes or constituents of lipid membranes within eukaryotic cells (Ikawa and Taylor, 1973; Treberg et al., 2006). MAs (trimethylamine (TMA), dimethylamine (DMA) and monomethylamine (MMA)) form part of a trace gas mix that is constantly emitted from the oceans and collectively these trace gases have major implications for the climate, largely through the production of particulate marine aerosols (Carpenter et al., 2012). Such aerosols can represent up to one-fifth of the total gaseous base compounds detected in the atmosphere over the oceans (Gibb et al., 1999a). Their global annual flux is estimated to be ~80 Gg per year and their production in surface seawater, and subsequent emission into the atmosphere, is thought to be largely driven by biotic processes (Ge et al., 2011). For example, over Cape Verde off the coast of West Africa, the accumulation of MAs in fine marine particles was positively correlated with algal blooms (Müller et al., 2009). The flux of MAs into the atmosphere is important as they can undergo a number of different reactions resulting in a complex set of effects on the climate. For instance, they can influence the absorption and scattering of ultraviolet radiation, the formation of cloud condensation nuclei (Ge et al., 2011), and the cloud droplet number concentration (Rinaldi et al., 2010). Moreover, off the coast of California, during periods of elevated primary production, a shift in the composition of secondary organic aerosols (SOAs) toward amine-derived compounds resulted in an increase in cloud condensation nuclei activity (Sorooshian et al., 2009). Thus, as a component of marine aerosols, MAs can actively affect the climate system.
Historically, the in situ quantification of MAs in the marine environment has proven challenging. Consequently, there are only a few studies reporting their standing stock concentrations (Carpenter et al., 2012). Generally, in surface seawater the concentration of MAs is in the nanomolar (nM) range, whereas in marine sediments it reaches low micromolar (μM) concentrations (Van Neste et al., 1987; Gibb et al., 1999b; Gibb and Hatton, 2004). Recent studies have identified a number of the key genes and enzymes catalyzing the degradation of TMA, TMAO and MMA in the marine environment (Chen et al., 2010, 2011; Lidbury et al., 2014) (Figure 1a). It is now known that bacteria capable of degrading MAs are abundant in surface seawater and are primarily related to the Alphaproteobacteria (Chen et al., 2011; Sun et al., 2011). Despite their low standing stock concentrations, expression of the key genes and enzymes catalyzing the degradation of MAs has been observed in surface seawater from various oceanic regions (Lidbury et al., 2014). Indeed, marine Alphaproteobacteria often heavily transcribe the TMAO-specific transporter suggesting that demethylation of TMAO to DMA may be a major process in surface ocean waters (Sowell et al., 2008; Ottesen et al., 2011, 2013; Williams et al., 2012; Gifford et al., 2013).

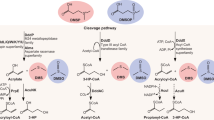
Scheme of (a) the proposed pathway of MA catabolism in R. pomeroyi DSS-3 and related MRC bacteria and (b) genomic regions encompassing the genes (dmmDABC) encoding the Dmm in R. pomeroyi DSS-3 and (c) Methylocella silvestris BL2. (d) Candidatus Pelagibacter ubique HTCC1062 does not possess dmmDABC in its genome despite containing all other genes required for TMA, TMAO and MMA degradation. amtB, ammonia transporter; CH2=H4F, 5,10-methylene tetrahydrofolate; DmmA, DmmB, DmmC, DmmD, DMA monooxygenase subunit A, B, C or D; GMA, gamma-glutamylmethylamide; GmaS, gamma-glutamylmethylamide synthetase; MgdABCD, N-methylglutamate dehydrogenase; MgsABC, N-methylglutamate synthase; NMG, N-methylglutamate; Tdm, trimethylamine N-oxide demethylase; Tmm, trimethylamine monooxygenase; tmoP, TMAO permease; TmoXVW, adenosine triphosphate-dependent TMAO transporter.
The marine Roseobacter clade (MRC) and SAR11 clade are two monophyletic groups of Alphaproteobacteria that use differing ecological strategies for growth (Luo et al., 2013). Both of these clades can catabolize MAs in order to generate reducing power, whereas the MRC can also utilize these compounds as a sole source of both carbon and nitrogen (Chen, 2012). Ruegeria pomeroyi DSS-3, a member of the MRC, has been used as a model organism to study the degradation of TMA, TMAO and MMA. However, how these marine bacteria degrade DMA remains unknown. In the methylotrophic soil bacterium Methylocella silvestris BL2, a three-gene cluster (dmmABC) is required for growth of this organism on DMA, as mutants lacking dmm genes ceased to grow on DMA as sole nitrogen source (Zhu et al., 2014). In addition, in another methylotrophic soil bacterium Paracoccus aminophilus JCM 7686, mutants lacking a functional dmmABC or an additional gene (dmmD), could no longer utilize DMA as a sole carbon source (Dziewit et al., 2015). Furthermore, a DMA monooxygenase (Dmm) has been purified from MA-grown Aminobacter aminovorans cells and shown to be a nicotinamide adenine dinucleotide phosphate-dependent enzyme that produces MMA and formaldehyde with DMA being the most active substrate (Alberta and Dawson, 1987). Dmm has a native molecular weight of ~210 kDa and comprises three subunits 42 000, 36 000 and 24 000 Da in size, each of which are essential for in vitro activity (Alberta and Dawson, 1987).
Here, we set out to determine the genes catalyzing DMA demethylation in marine bacteria using R. pomeroyi DSS-3 as the model organism. Dmm was heterologously expressed in Escherichia coli and the function of the predicted three-gene cluster, dmmABC, was confirmed for the first time by enzymatic, chemical and growth assays. We also demonstrate that, unlike the genes required for the catabolism of TMA, TMAO and MMA, the genes required for DMA catabolism are absent from key marine bacterial taxa and are subsequently depleted in metagenomes derived from oceanic surface waters.
Materials and methods
Bacterial cultivation
The strains used in this study are listed in Supplementary Table S1. R. pomeroyi wild-type and mutants were grown in a marine ammonium minimal salts medium (Thompson et al., 1995) with slight modifications (Lidbury et al., 2015) using 10 mM glucose as carbon source. TMA, TMAO, DMA and MMA (1 mM) were added as sole nitrogen source. To observe growth on different nitrogen sources, cultures (n=3) were set up in 125 ml serum vials containing 25 ml medium. Overnight starter cultures were harvested by centrifugation (1500 × g, 5 min) and washed three times in nitrogen-free marine ammonium minimal salts before inoculation (8% v/v). Cultures were kept under constant agitation (150 r.p.m.) at 30 °C.
Overexpression of dmmABC and dmmDABC in a heterologous host
All primers used in this study are listed in Supplementary Table S2. Either dmmABC encoding the structural components of Dmm or the entire operon dmmDABC were subcloned into the pGEM-T EASY vector (Promega, Southampton, UK). Sequence integrity was checked before digestion using the restriction enzymes NheI and HindIII and subsequent ligation into the expression vector pET28a, which was transformed into E. coli BLR(DE3)pLysS (Promega). Transformed E. coli cells were grown for 32 h at 25 °C in the presence of 0.2 mM isopropyl β-D-1-thiogalactopyranoside and 1 mM DMA.
Mutagenic analysis and mutant complementation in R. pomeroyi
A dmmD disrupted mutant (dmmD::Gm) in R. pomeroyi DSS-3 was constructed by cloning part of the gene (Spo1579) into the pGEM-T EASY vector. A gentamicin resistance cassette (Dennis and Zylstra, 1998) was inserted into a naturally occurring SpeI site located near the centre of the gene. The mutated construct was cloned into the suicide vector, pk18mobsacB (Schäfer et al., 1994), and mobilized into R. pomeroyi via conjugation with E. coli S17-1 electrocompetent cells. Transconjugants were streaked onto gentamicin plates containing MMA as the sole nitrogen source to counterselect against E. coli (Lidbury et al., 2014). Double homologous recombination events were selected for by transconjugant sensitivity to kanamycin. The mutation was confirmed by PCR and sequencing.
To complement the dmmD::Gm with dmmDABC plus its native promoter, the entire gene cluster was amplified introducing the restriction sites XbaI and KpnI at the 5′ and 3′ ends, respectively. For complementation with the structural genes dmmABC, the promoter alone was amplified introducing the restriction sites XbaI and HindIII at the 5′ and 3′ ends, respectively. In addition, dmmABC was amplified introducing the restriction sites HindIII and KpnI at the 5′ and 3′ ends, respectively. For complementation using just dmmD, this gene (Spo1579) plus the promoter were amplified introducing the restriction sites BamHI and HindIII at the 5′ and 3′ end, respectively. All PCR fragments were subcloned into the pGEM-T EASY vector. Sequence integrity was checked before cloning the construct into the broad-host range plasmid pBBR1MCS-km (Kovach et al., 1995) and mobilized into dmmD::Gm via conjugation as before. Transconjugants were selected by growth on half-strength Yeast Tryptone Sea Salts (1/2 YTSS) media containing 80 μg ml−1 kanamycin and 10 μg ml−1 gentamicin. Complementation was confirmed by PCR and sequencing.
Quantification of MAs
Cells were boiled for ⩾10 min and debris was removed via centrifugation (17 000 × g, 5 min). TMA, TMAO, DMA and MMA were quantified on a cation-exchange ion chromatograph (881 Compact IC pro, Metrohm, Runcorn, UK) supplied with Metrosep C 4 guard (Metrohm, Switzerland) and Metrosep C 4-250/4.0 separation column, and a conductivity detector (Metrohm, Switzerland) using an external calibration (Lidbury et al., 2014).
Analysis of enzymes involved in MA metabolism in sequenced marine microbial genomes
Single amplified genomes used in this study derived from the Integrated Microbial Genome (IMG) database of the Joint Genome Institute (JGI) (https://img.jgi.doe.gov/cgi-bin/m/main.cgi). All available defined marine bacterial genomes were screened for enzymes catalyzing MA degradation using a BLASTP analysis with Tmm (Spo1551), Tdm (Spo1562), DmmD (Spo1579), DmmA (Spo1580), DmmB (Spo1581), DmmC (Spo1582), GmaS (Spo1573) and TmoX (Spo1548) from R. pomeroyi DSS-3 as query sequences using a stringent cutoff value of e-50. Marine bacterial genomes containing genes encoding these proteins are listed in Supplementary Table S3. Taxonomy information at the phylum, class and order level was exported from the IMG/JGI database. For phylogenetic analysis, amino-acid sequences of dmmD, dmmA, dmmB and dmmC from 36 taxa were aligned individually by MUSCLE (Edgar, 2004), trimmed at either end and combined to one alignment. Evolutionary analysis was conducted in MEGA7 (Kumar et al., 2016) on a total of 1043 positions remaining in the data set after exclusion of gaps and missing data. A phylogenetic tree was inferred by a maximum likelihood approach applying the WAG model (Whelan and Goldman, 2001) with 999 bootstrap replicates and using a maximum parsimony tree derived from neighbor-joining as the initial tree.
Analysis of enzymes involved in MA metabolism in marine metagenomes and metatranscriptomes
The metagenomes used in this study and the abundances of MA degradation genes are listed in Supplementary Table S4. Metagenomes were chosen from the IMG/JGI database and predominantly consisted of sites used in the global ocean sampling expedition (Rusch et al., 2007). A BLASTP analysis was performed using a stringency of >30% identity and a cutoff value of e-50. Query sequences were identical to those described above. The number of retrieved sequences for each protein was normalized by dividing the length of the query by the length of RecA. Finally, the normalized hits were divided by the number of hits retrieved for two single copy genes (recA and gyrB) to obtain the percentage of MA-utilizing marine bacteria present at each site. For phylogenetic analysis, hits were clustered using CD-HIT (Huang et al., 2010) at a similarity cutoff of 0.8. Representative sequences were then used as query in BLASTP (multiple query function) searches using the National Centre for Bioinformatics database (nr). The best hit was used to assign taxonomy at the family level.
The metatranscriptomes used in this study are listed in Supplementary Table S5. Metatranscriptomes deposited in the IMG/JGI database were used for a BLASTP analysis with a stringency level of >40% similarity and a cutoff value of e-20. Query sequences were identical to those used above and data normalized by the length of RecA as described above.
Results
Identification of a four-gene cluster in R. pomeroyi DSS-3
R. pomeroyi can utilize TMA, DMA and MMA as a sole nitrogen source (Lidbury et al., 2015). Therefore, a BLASTP analysis on R. pomeroyi was performed to identify candidate genes involved in DMA catabolism using the three-gene cluster identified as dmmABC (Msil_3607, Msil_3608 and Msil_3609) from M. silvestris as the query sequences (Zhu et al., 2014). Three open reading frames, Spo1580, Spo1581 and Spo1582 shared good homology with Msil_3607 (E-value, 4.0e-32; 38.92%), Msil_3608 (E-value, 4.0e-75; 41.07%) and Msil_3609 (E-value, 420e-157; 62.24%), respectively (Figures 1b and c). Another open reading frame, Spo1579, found in an apparent operon with the other three open reading frames, shared homology with Msil_3605 (E-value, 2.0e-67; 35.75%), both of which contain a conserved tetrahydrofolate (H4F)-binding domain (GcvT). The GcvT domain is highly conserved in DmmD homologs (Zhu et al., 2014) and is also found in bacterial TMAO demethylase (Tdm) (Lidbury et al., 2014). Spo1579, Spo1580, Spo1581 and Spo1582 are hereafter referred to as dmmD, dmmA, dmmB and dmmC, respectively. Unlike in M. silvestris, dmmD was always co-located with dmmABC in the genomes of various MRC isolates screened (Supplementary Table S3), suggesting that its expression is tightly coordinated to that of dmmABC. Interestingly, dmmDABC was absent from the genome of Candidatus Pelagibacter ubique HTCC1062 (Figure 1d), a member of the SAR11 clade that can utilize TMA and MMA (Sun et al., 2011).
DmmABC forms a functional Dmm
To determine if all four subunits of Dmm were essential for DMA demethylation, both dmmDABC and dmmABC were cloned into the expression vector pET28a and transformed into E. coli BLR(DE3)pLysS. In the E. coli strain harboring dmmABC, complete degradation of DMA (1 mM) occurred within 8 h while the concentration of MMA increased in a stoichiometric 1:1 manner (Figure 2). In the E. coli strain harboring dmmDABC, DMA degradation in accordance with MMA production still occurred, albeit at a slower rate, again, stoichiometrically in a 1:1 ratio (Figure 2). In cultures complemented with the empty pET28a vector, no DMA degradation and thus no MMA production was observed (Figure 2), whereas cultures grew comparably (Supplementary Figure S1). Together, these results show that the three-subunit cluster alone forms a functional Dmm.

Assessment of (a) DMA degradation and (b) MMA accumulation in recombinant E. coli following heterologous expression of either the complete dmmDABC gene cluster from R. pomeroyi or just the structural genes (+ dmmABC), or of the expression vector pET28a as a negative control (C-). Results presented are the mean of triplicates and error bars denote s.d.
dmmD is essential for growth on DMA and other MAs in R. pomeroyi
To determine the function of dmmD in R. pomeroyi, the gene was disrupted by insertion of a gentamicin resistance marker (Dennis and Zylstra, 1998) and the dmmD::Gm mutant subsequently grown on MAs including DMA as a sole nitrogen source. Disruption of the dmmD gene resulted in an inability of the mutant to grow on TMA, TMAO or DMA as a sole nitrogen source (Figures 3a–c). However, growth on MMA and NH4+ was unaffected (Figure 3d,Supplementary Figure S2a). Complementation with dmmD did not restore growth in comparison with the wild type (Figures 3a–c). dmmDABC forms a single operon and therefore deletion of dmmD may have affected the downstream expression of dmmABC. When grown on TMA and TMAO, the dmmD mutant accumulated DMA in the culture medium revealing a bottleneck in the MA degradation pathway (Supplementary Figure S3). However, when grown on DMA as the sole nitrogen source DMA degradation was slightly enhanced by complementation (Figure 3c), suggesting that dmmD may be required for DMA degradation in R. pomeroyi.

Growth of R. pomeroyi DSS-3 wild-type (WT), dmmD mutant (dmmD::Gm) and its complementation with dmmD (dmmD::Gm+dmmD) on (a) TMA, (b) TMAO, (c) DMA and (d) MMA as the sole nitrogen source. Solid lines represent cell growth. Dashed lines represent the degradation of the appropriate substrate with the concentrations of TMA, TMAO, DMA and MMA being quantified throughout the whole experiment. Results presented are the mean of triplicates and error bars denote s.d.
Owing to the potential polar effect on dmmABC by deletion of dmmD, the dmmD mutant was complemented with either the four-gene cluster dmmDABC (dmmD::Gm+dmmDABC) or the three subunits of Dmm, that is, dmmABC (dmmD::Gm+dmmABC). To achieve this, these two gene clusters were cloned into the broad-host range plasmid pBBR1MCS-km (Kovach et al., 1995) together with the putative promoter located at the 5′ untranslated region upstream of dmmD. For the dmmD::Gm+dmmDABC complemented mutant, growth on TMA and TMAO as a sole nitrogen source was restored while for dmmD::Gm+dmmABC, missing an intact dmmD, the complemented mutant failed to grow on either TMA or TMAO (Figures 4a and b). Consequently, in the dmmD::Gm+dmmABC complemented mutant, DMA accumulated in the medium as TMA or TMAO degradation occurred (Supplementary Figure S4). However, both complemented strains could degrade and subsequently grow on DMA, MMA and NH4+ as sole nitrogen sources (Figures 4c and d, Supplementary Figure S5a), suggesting that dmmD is essential for TMA and TMAO degradation but not for growth on DMA or MMA in this bacterium.

Growth of R. pomeroyi DSS-3 wild-type (WT), and the dmmD mutant (dmmD::Gm) complemented with either the four-gene cluster dmmDABC (dmmD::Gm+dmmDABC) or only the structural genes dmmABC (dmmD::Gm+dmmABC) along with the promoter on different nitrogen sources. Nitrogen was supplied in the form of (a) TMA, (b) TMAO, (c) DMA and (d) MMA. Solid lines represent cell growth. Dashed lines represent the degradation of the appropriate substrate with the concentrations of TMA, TMAO, DMA and MMA being quantified throughout the whole experiment. Results presented are the mean of triplicates and error bars denote s.d.
The distribution of DmmDABC in marine bacterial genomes and metagenomes
The distribution of genes encoding DmmDABC was investigated using BLASTP analysis among marine bacterial genomes deposited in the IMG database of the JGI. In parallel, the distribution of genes encoding the other enzymes required for growth on MAs (for example, Tmm, Tdm, TmoX and GmaS) was also determined using R. pomeroyi homologs as the query sequences. The dmmDABC gene cluster was identified in 30 isolates related to Alphaproteobacteria and 6 related to Gammaproteobacteria (Figures 5a). The majority of Alphaproteobacteria homologs were related to the MRC (27/30). In addition, dmmDABC homologs were retrieved from Candidatus Puniceispirillum marinum IMCC1322 (IMCC1132), a member of the cosmopolitan SAR116 clade (Oh et al., 2010; Giovannoni and Vergin, 2012) and clustered with the MRC homologs suggesting horizontal gene transfer has occurred (Figure 5a). A number of dmmDABC homologs were also found in the genomes of largely uncultivated pelagic Roseobacter (Figure 5a,Supplementary Table S3), some of which have been previously reported to possess features of a free-living life style (for example, Rhodobacterales sp. HTCC2255) (Billerbeck et al., 2016; Zhang et al., 2016). Notably, all representatives of the Pelagibacterales (SAR11 clade) lack homologs of the genes encoding DmmDABC (Figure 5, Supplementary Table S3), whereas genes encoding GmaS, Tmm, Tdm and TmoX were ubiquitous within the genomes of strains related to this clade (Figure 5b,Supplementary Table S3).

Distribution of genes for MA metabolism in marine bacterial isolates. (a) Maximum likelihood phylogenetic tree of dmmDABC homologs in marine bacterial isolates. For each node bootstrap values (999 replicates) >50% are given. MRC are marked in orange. An asterisk indicates pelagic Roseobacter, with the affiliation of two representatives to the largely uncultivated pelagic Roseobacter lineages according to Zhang et al. (2016) given in brackets. (b) Phylogenetic distribution of the genes encoding the enzymes involved in MA metabolism. TmoX, substrate-binding protein of the TMAO transporter, other abbreviations are as described in Figure 1.
Previous studies have shown that tmm, tdm and gmaS are abundant in marine metagenomes primarily because of their occurrence in SAR11 clade bacteria (Chen et al., 2011; Lidbury et al., 2014). We hypothesized that the abundance of dmmDABC in marine metagenomes would be lower than that of tmm, tdm and gmaS, reflecting their absence from the genomes of SAR11 clade bacteria. To test this hypothesis, a number of metagenomes deposited in the IMG/JGI database, predominantly from the global ocean sampling expedition (Rusch et al., 2007) were screened (stringency, e-50) for the presence of dmmDABC as well as tmm, tdm, tmoX and gmaS using the R. pomeroyi homologs as the query sequences. To determine the percentage of marine bacteria possessing MA degradation genes present at each site, counts were normalized against the average counts of two single copy genes (recA and gyrB). As expected tmm, tdm, tmoX and gmaS were present in 20–25% of marine bacteria (Figure 6a). However, dmmDABC was found at a much lower abundance (Figure 6a, Supplementary Table S4). To rule out the possibility that the under-representation of dmmDABC genes in marine metagenomes was because of the use of a high stringency cutoff value (e-50), we re-analyzed metagenomes from the global ocean sampling data set with a range of stringency thresholds (e-40, e-20, e-10, e-8) and the number of hits related to dmmDABC did not increase relative to that of tmm and tdm (Supplementary Figure S6). dmmDABC were also retrieved from metagenomes associated with high primary productivity, for example, a photosynthetic picoeukaryote bloom in the Norwegian Sea (IMG genome ID 3300002186), albeit at a lower abundance than other MA-degrading genes (Supplementary Table S4). Phylogenetic analysis revealed that dmmDABC sequences retrieved from marine metagenomes were primarily related to the MRC (Figure 6b). It should be noted that several tmm and tdm sequences were related to the newly identified gammaproteobacterium, Candidatus Thioglobus singularis (Marshall and Morris, 2015). A similar pattern was also observed when scrutinizing metatranscriptomes (Supplementary Table S5). No transcripts related to dmmDABC could be detected from various open ocean and coastal ocean waters, whereas transcripts related to various other genes involved in the MA degradation pathway (tmm, tdm, gmaS or tmoX) were readily detected (Supplementary Table S5, Ottesen et al., 2011, 2013; Gifford et al., 2013).

Distribution of genes encoding proteins for MA metabolism in selected marine metagenomes. (a) Abundance of selected genes in marine bacteria, and (b) their phylogenetic affiliation. In the box-whisker plot whiskers represent the 5 and 95 percentiles and the line corresponds to the median. Circles represent outliers with all high-range outliers of Tmm, Tdm, GmaS and TmoX deriving from the same Sargasso Sea metagenome sample (Supplementary Table S4). The phylogenetic composition represents the normalized relative abundances of MA-degrading genes using metagenomes primarily retrieved from the global ocean sampling (GOS) data set (Rusch et al., 2007) (see Supplementary Table S4). Abbreviations are as described in Figure 1.
Discussion
Recently, the genes involved in DMA degradation were identified in methylotrophic soil bacteria (Zhu et al., 2014; Dziewit et al., 2015). However, neither study conclusively demonstrated the functionality of Dmm at the protein level. By identifying R. pomeroyi dmmDABC homologs similar to those found in M. silvestris and P. aminophilus we were able to confirm that dmmABC does indeed encode a functional Dmm, an enzyme originally described in A. aminovorans (Alberta and Dawson, 1987). In both M. silvestris and P. aminophilus, dmmD was not essential for growth on MAs, but disruption of this gene did affect their growth rates on TMA, DMA and TMAO (the latter substrate was shown for M. silvestris only) (Zhu et al., 2014; Dziewit et al., 2015). These findings, alongside the data presented here (Figures 2–4), further suggest that dmmD is required for normal growth on MAs. As DmmD possesses a H4F-binding domain, its primary role is likely to be involved in the conjugation of free formaldehyde, released from the demethylation of DMA, with the one carbon (C1) carrier molecule H4F (Zhu et al., 2014). Unlike M. silvestris and P. aminophilus, marine bacteria only possess the genes for C1 oxidation via the H4F pathway, lacking the genes required for C1 oxidation through either the tetrahydromethanopterin (H4MPT), glutathione-linked pathway or the formaldehyde-activating enzyme (Chistoserdova, 2011; Dziewit et al., 2015). Thus, there is a greater dependency of the H4F-linked C1 oxidation pathway to deal with formaldehyde stress. The consistently tight genetic arrangement of dmmDABC in marine bacteria coupled with the non-essential function of dmmD in DMA or MMA degradation further strengthens the hypothesis that dmmD serves a key role in reducing formaldehyde toxicity. Furthermore, conjugation with H4F also allows the C1 unit to be fully oxidized to CO2 while generating reducing power (Lidbury et al., 2015).
The absence of dmmDABC from members of the SAR11 clade as well as abundant marine Gammaproteobacteria and Deltaproteobacteria is intriguing. C. Pelagibacter ubique HTCC1062 has been shown to oxidize TMA, TMAO and MMA in order to generate adenosine triphosphate (Sun et al., 2011). However, currently there is no evidence that this bacterium or any other member of the SAR11 clade can oxidize DMA. Furthermore, there is no evidence that SAR11 clade bacteria can grow on MAs as a source of nitrogen, which would require the complete demethylation of MAs, including DMA (Lidbury et al., 2015). During N-limitation, C. Pelagibacter ubique HTCC1062 does express a protein that is predicted to be a general amine oxidase (Smith et al., 2013), but its role in DMA oxidation has not been confirmed experimentally. In contrast to the Pelagibacterales, dmmDABC is found in pelagic Roseobacters (Figure 5a, Supplementary Table S3), thus, ruling out an affiliation of its absence with a pelagic life style. Representatives possessing the dmm genes have been found in the streamlined, largely non-cultivated pelagic Roseobacter lineages DC5-80-3 and NAC11-7 (Zhang et al., 2016), whereas the other globally abundant pelagic Roseobacter CHAB-I-5 lineage (Billerbeck et al., 2016; Zhang et al., 2016) only shows genetic evidence for oxidation of TMA, TMAO and MMA, but not DMA (that is, no dmm genes found in their genomes).
The flux of MAs from surface seawaters is important as these compounds can lead to the formation of aerosols and thus cloud condensation nuclei (Ge et al., 2011). Owing to the scarcity of labile organic nitrogen in marine surface waters, biological consumption of MAs as a nitrogen source is likely to be a major limitation on the air–sea exchange of these compounds (Balch, 1985; Carpenter et al., 2012; Chen, 2012). In addition, R. pomeroyi and C. Pelagibacter ubique rapidly turn over MAs as an energy source (Sun et al., 2011; Lidbury et al., 2015), further reducing the amount of MAs available for air–sea exchange. The lack of dmmDABC homologs relative to other MA degradation genes (tmm, tdmand gmaS) in marine metagenomes suggests that DMA may accumulate in surface waters and therefore be susceptible to a greater amount of air–sea exchange. In support of this hypothesis, besides methanesulfonic acid, DMA amine salts were often the most abundant SOAs detected in fine marine particles at sites located in the North and Tropical Atlantic Ocean (Facchini et al., 2008; Müller et al., 2009). In these studies, a link between elevated concentrations of amine-derived SOAs detected in fine marine particles and elevated levels of primary production was observed and thought to be of biological origin. In another study, a shift toward amine-derived SOAs and the subsequent accumulation of cloud condensation nuclei was correlated with elevated periods of primary production (Sorooshian et al., 2009). In this context, metagenomic data collected during a photosynthetic picoeukaryote bloom in the Norwegian Sea revealed that dmmDABC homologs were substantially reduced (5.95% of total bacteria) compared with those of tmm, tdm and gmaS (42.83% of total bacteria) (Supplementary Table S4). Similarly, in the North Sea where members of the MRC are often numerically abundant during phytoplankton blooms (Teeling et al., 2012; Wemheuer et al., 2015), dmmDABC homologs were again under-represented (6% of total bacteria) relative to other MA degradation genes (21% of total bacteria) (an average of 41 metagenomes, Supplementary Table S4). Therefore, a lack of DMA-degrading bacteria relative to other MA-degrading bacteria in the euphotic zone, especially during periods of elevated primary production, may be an explanation for the higher abundance of DMA-containing SOAs.
In conclusion, this study has confirmed the genes and enzyme catalyzing DMA degradation in marine bacteria and revealed a potential bottleneck in the MA degradation pathway in surface seawaters. We propose that this metabolic bottleneck likely explains the elevated abundance of DMA-derived amine salts detected in fine marine particles. Further research on the environmental cycling of MAs, especially DMA, is required to better understand the air-sea exchange of these climatically important compounds.
References
Alberta JA, Dawson JH .(1987). Purification to homogeneity and initial physical characterization of secondary amine monooxygenase. J Biol Chem 262: 11857–11863.
Balch WM .(1985). Lack of an effect of light on methylamine uptake by phytoplankton. Limnol Oceanogr 30: 665–674.
Billerbeck S, Wemheuer B, Voget S, Poehlein A, Giebel H-A, Brinkhoff T et al.(2016). Biogeography and environmental genomics of the Roseobacter-affiliated pelagic CHAB-I-5 lineage. Nat Microbiol 1: 16063.
Carpenter LJ, Archer SD, Beale R .(2012). Ocean-atmosphere trace gas exchange. Chem Soc Rev 41: 6473–6506.
Chen Y, McAleer KL, Murrell JC .(2010). Monomethylamine as a nitrogen source for a non-methylotrophic bacterium Agrobacterium tumefaciens. Appl Environ Microbiol 76: 4102–4104.
Chen Y, Patel NA, Crombie A, Scrivens JH, Murrell JC .(2011). Bacterial flavin-containing monooxygenase is trimethylamine monooxygenase. Proc Natl Acad Sci USA 108: 17791–17796.
Chen Y .(2012). Comparative genomics of methylated amine utilization by marine Roseobacter clade bacteria and development of functional gene markers (tmm gmaS. Environ Microbiol 14: 2308–2322.
Chistoserdova L .(2011). Modularity of methylotrophy, revisited. Environ Microbiol 13: 2603–2622.
Dennis JJ, Zylstra GJ .(1998). Plasposons: modular self-cloning mini-transposon derivatives for rapid genetic analysis of gram-negative bacterial genomes. Appl Environ Microbiol 64: 2710–2715.
Dziewit L, Czarnecki J, Prochwicz E, Wibberg D, Schlüter A, Pühler A et al.(2015). Genome-guided insight into the methylotrophy of Paracoccus aminophilus JCM 7686. Front Microbiol 6: 852.
Edgar RC .(2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797.
Facchini MC, Decesari S, Rinaldi M, Carbone C, Finessi E, Mircea M et al.(2008). Important source of marine secondary organic aerosol from biogenic amines. Environ Sci Technol 42: 9116–9121.
Ge X, Wexler AS, Clegg SL .(2011). Atmospheric amines – part I. A review. Atmos Environ 45: 524–546.
Gibb SW, Mantoura RFC, Liss PS .(1999a). Ocean-atmosphere exchange and atmospheric speciation of ammonia and methylamines in the region of the NW Arabian Sea. Global Biogeochem Cycles 13: 161–178.
Gibb SW, Mantoura RFC, Liss PS, Barlow RG .(1999b). Distributions and biogeochemistries of methylamines and ammonium in the Arabian Sea. Deep-Sea Res Pt II 46: 593–615.
Gibb SW, Hatton AD .(2004). The occurrence and distribution of trimethylamine-N-oxide in Antarctic coastal waters. Mar Chem 91: 65–75.
Gifford SM, Sharma S, Booth M, Moran MA .(2013). Expression patterns reveal niche diversification in a marine microbial assemblage. ISME J 7: 281–298.
Giovannoni SJ, Vergin KL .(2012). Seasonality in ocean microbial communities. Science 335: 671–676.
Huang Y, Niu B, Gao Y, Fu L, Li W .(2010). CD-HIT suite: a web server for clustering and comparing biological sequences. Bioinformatics 26: 680–682.
Ikawa M, Taylor RF .(1973) Choline and related substances in algae. In: Martin, Padilla G (eds). Marine Pharmacognosy: Action of Marine Biotoxins at the Cellular Level. Academic Press INC.: New York, NY, USA, pp 203–236.
Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM II et al.(1995). Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166: 175–176.
Kumar S, Stecher G, Tamura K .(2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33: 1870–1874.
Lidbury I, Murrell JC, Chen Y .(2014). Trimethylamine N-oxide metabolism by abundant marine heterotrophic bacteria. Proc Natl Acad Sci USA 111: 2710–2715.
Lidbury IDEA, Murrell JC, Chen Y .(2015). Trimethylamine and trimethylamine N-oxide are supplementary energy sources for a marine heterotrophic bacterium: implications for marine carbon and nitrogen cycling. ISME J 9: 760–769.
Luo H Csuros M, Hughes AL, Moran MA .(2013).Evolution of divergent life history strategies in marine alphaproteobacteria. mBio 4: pii: e00373–13.
Marshall KT, Morris RM .(2015). Genome sequence of ‘Candidatus Thioglobus singularis’ strain PS1, a mixotroph from the SUP05 clade of marine Gammaproteobacteria. Genome Announc 3: e01155–01115.
Müller C, Iinuma Y, Karstensen J, van Pinxteren D, Lehmann S, Gnauk T et al.(2009). Seasonal variation of aliphatic amines in marine sub-micrometer particles at the Cape Verde islands. Atmos Chem Phys 9: 9587–9597.
Oh H-M, Kwon KK, Kang I, Kang SG, Lee J-H, Kim S-J et al.(2010). Complete genome sequence of ‘Candidatus Puniceispirillum marinum’ IMCC1322, a representative of the SAR116 Clade in the Alphaproteobacteria. J Bacteriol 192: 3240–3241.
Ottesen EA, Marin R III, Preston CM, Young CR, Ryan JP, Scholin CA et al.(2011). Metatranscriptomic analysis of autonomously collected and preserved marine bacterioplankton. ISME J 5: 1881–1895.
Ottesen EA, Young CR, Eppley JM, Ryan JP, Chavez FP, Scholin CA et al.(2013). Pattern and synchrony of gene expression among sympatric marine microbial populations. Proc Natl Acad Sci USA 110: E488–E497.
Rinaldi M, Decesari S, Finessi E, Giulianelli L, Carbone C, Fuzzi S et al.(2010). Primary and secondary organic marine aerosol and oceanic biological activity: recent results and new perspectives for future studies. Adv Meteorol 2010: doi:10.1155/2010/310682.
Rusch DB, Halpern AL, Sutton G, Heidelberg KB, Williamson S, Yooseph S et al.(2007). The Sorcerer II global ocean sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biol 5: e77.
Schäfer A, Tauch A, Jäger W, Kalinowski J, Thierbach G, Pühler A .(1994). Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145: 69–73.
Smith DP, Thrash JC, Nicora CD, Lipton MS, Burnum-Johnson KE, Carini P et al.(2013). Proteomic and transcriptomic analyses of ‘Candidatus Pelagibacter ubique’ describe the first PII-independent response to nitrogen limitation in a free-living alphaproteobacterium. mBio 4: e00133–12.
Sorooshian A, Padró LT, Nenes A, Feingold G, McComiskey A, Hersey SP et al.(2009). On the link between ocean biota emissions, aerosol, and maritime clouds: airborne, ground, and satellite measurements off the coast of California. Global Biogeochem Cycles 23: GB4007.
Sowell SM, Wilhelm LJ, Norbeck AD, Lipton MS, Nicora CD, Barofsky DF et al.(2008). Transport functions dominate the SAR11 metaproteome at low-nutrient extremes in the Sargasso Sea. ISME J 3: 93–105.
Sun J, Steindler L, Thrash JC, Halsey KH, Smith DP, Carter AE et al.(2011). One carbon metabolism in SAR11 pelagic marine bacteria. PLoS One 6: e23973.
Teeling H, Fuchs BM, Becher D, Klockow C, Gardebrecht A, Bennke CM et al.(2012). Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336: 608–611.
Thompson AS, Owens N, Murrell JC .(1995). Isolation and characterization of methanesulfonic acid-degrading bacteria from the marine environment. Appl Environ Microbiol 61: 2388–2393.
Treberg JR, Speers-Roesch B, Piermarini PM, Ip YK, Ballantyne JS, Driedzic WR .(2006). The accumulation of methylamine counteracting solutes in elasmobranchs with differing levels of urea: a comparison of marine and freshwater species. J Exp Biol 209: 860–870.
Van Neste A, Duce RA, Lee C .(1987). Methylamines in the marine atmosphere. Geophys Res Lett 14: 711–714.
Wemheuer B, Wemheuer F, Hollensteiner J, Meyer F-D, Voget S, Daniel R .(2015). The green impact: bacterioplankton response towards a phytoplankton spring bloom in the southern North Sea assessed by comparative metagenomic and metatranscriptomic approaches. Front Microbiol 6: 805.
Whelan S, Goldman N .(2001). A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol 18: 691–699.
Williams TJ, Long E, Evans F, DeMaere MZ, Lauro FM, Raftery MJ et al.(2012). A metaproteomic assessment of winter and summer bacterioplankton from Antarctic Peninsula coastal surface waters. ISME J 6: 1883–1900.
Zhang Y, Sun Y, Jiao N, Stepanauskas R, Luo H .(2016). Ecological genomics of the uncultivated marine Roseobacter lineage CHAB-I-5. Appl Environ Microbiol 82: 2100–2111.
Zhu Y, Jameson E, Parslow RA, Lidbury I, Fu T, Dafforn TR et al.(2014). Identification and characterization of trimethylamine N-oxide (TMAO) demethylase and TMAO permease in Methylocella silvestris BL2. Environ Microbiol 16: 3318–3330.
Acknowledgements
This project was funded by Natural Environment Research Council (NERC) grant NE/M002233/1. We thank Mr Zijing Cao, University of Warwick, who helped with the construction of the dmmD::Gm mutant used in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Lidbury, I., Mausz, M., Scanlan, D. et al. Identification of dimethylamine monooxygenase in marine bacteria reveals a metabolic bottleneck in the methylated amine degradation pathway. ISME J 11, 1592–1601 (2017). https://doi.org/10.1038/ismej.2017.31
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2017.31
This article is cited by
-
Genomic profiling and characteristics of a C1 degrading heterotrophic fresh-water bacterium Paracoccus sp. strain DMF
Archives of Microbiology (2024)
-
Assessment of prokaryotic communities in Southwestern Atlantic deep-sea sediments reveals prevalent methanol-oxidising Methylomirabilales
Scientific Reports (2023)
-
Insight into the metabolic pathways of Paracoccus sp. strain DMF: a non-marine halotolerant methylotroph capable of degrading aliphatic amines/amides
Environmental Science and Pollution Research (2023)
