
Abstract
To date, very little is known about the bacterial core community of marine sediments. Here we study the environmental distribution, abundance and ecogenomics of the gammaproteobacterial Woeseiaceae/JTB255 marine benthic group. A meta-analysis of published work shows that the Woeseiaceae/JTB255 are ubiquitous and consistently rank among the most abundant 16S rRNA gene sequences in diverse marine sediments. They account for up to 22% of bacterial amplicons and 6% of total cell counts in European and Australian coastal sediments. The analysis of a single-cell genome, metagenomic bins and the genome of the next cultured relative Woeseia oceani indicated a broad physiological range, including heterotrophy and facultative autotrophy. All tested (meta)genomes encode a truncated denitrification pathway to nitrous oxide. The broad range of energy-yielding metabolisms possibly explains the ubiquity and high abundance of Woeseiaceae/JTB255 in marine sediments, where they carry out diverse, but yet unknown ecological functions.
Similar content being viewed by others


Introduction
Marine sediments are hot spots of microbially catalyzed element cycling and are Earth's major carbon sink (Burdige, 2007). While there is consensus that some bacterial classes such as Flavobacteria, Gamma- and Deltaproteobacteria generally thrive in marine sediments (Amaral-Zettler et al., 2010; Orcutt et al., 2011; Zinger et al., 2011), it is still not known whether bacterial lineages of lower taxonomic ranks belong to the ‘core microbiome’ of marine sediments. Recently, Bienhold et al. (2016) identified representatives of the gammaproteobacterial JTB255-marine benthic group (MBG) as ubiquitous core members of microbial communities in abyssal and bathyal surface sediments. Consistent with this, we identified the JTB255-MBG being among the dominant bacterial groups in 13 coastal sediments across Europe and Australia (Dyksma et al., 2016). In some of these sediments, members of the JTB255-MBG assimilated inorganic carbon (Dyksma et al., 2016), which would be in line with a hypothesized sulfur oxidation potential in this group (Bowman et al., 2005). However, the genomic repertoire of this group is still unknown.
In this study, we surveyed published the next-generation-sequencing (NGS) datasets of 16S rRNA gene amplicons from marine sediments and determined relative JTB255-MBG cell abundances in five tidal marine sediments to underscore their environmental importance. By analyzing a single amplified genome (SAG), metagenomic bins from two distant sites and the genome of the recently isolated, cultured and closely related Woeseia oceani (Du et al., 2016), we explored the genetic and metabolic potential of this group. Using 16S rRNA gene sequences and concatenated ribosomal proteins, we revisit the phylogenetic affiliation of the JTB255-MBG, indicating an affiliation with the Woeseiaceae.
Distribution and environmental importance in marine sediments
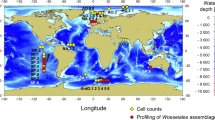
To assess the relative abundance of 16S rRNA gene sequences of JTB255-MBG in marine sediments, we surveyed recently published 16S rRNA gene amplicon studies that provided a sufficient taxonomic resolution along with relative sequence abundances (Figure 1). The JTB255-MBG occurs in high relative sequence abundances in all types of marine benthic habitats, including hydrothermal sites, coastal and deep-sea, and organic-rich and -poor sediments (Supplementary Figure S1, Supplementary Table S1). Intriguingly, among the lower taxonomic ranks (family to genus), the JTB255-MBG constituted the group with the highest relative sequence abundance at 32 sites (Figure 1, for example, Bowman et al., 2005; Schauer et al., 2010; Ruff et al., 2014; Liu et al., 2015). At 30 sites, the JTB255-MBG ranked among the top 10 sequence groups (for example, Tully and Heidelberg, 2013; Liu et al., 2015). It was found in all tested NGS-amplicon datasets. In 13 selected sediment samples surveyed by Dyksma et al., 2016, they accounted for an average of 9.8% of all bacterial sequences (Supplementary Table S2), which is a percentage close to their average relative cell abundance of 5.0% in some sediments as was determined by fluorescence in situ hybridization (Figure 1, Supplementary Table S2, Supplementary Figure S7).

Global distribution and relative sequence abundances of 16S rRNA genes of the Woeseiaceae/JTB255 in marine benthic habitats. Color-coded circles refer to the relative sequence abundance of Woeseiaceae/JTB255 in NGS amplicon or clone studies according to SILVAngs (Quast et al., 2013). Blue circles refer to the only qualitative detection of sequences of the Woeseiaceae/JTB255 in clone studies. Note that in previous publications, using outdated training sets for amplicon classification, the 16S rRNA gene sequences of the Woeseiaceae/JTB255 have often been classified as marine Sinobacteraceae/Xanthomonadales. An annotated map with references is given in the Supplementary Information (Supplementary Figure S1, Supplementary Table S1).
To study the distribution patterns of JTB255-MBG populations in eight tidal sediments from Europe and Australia (Dyksma et al., 2016), we performed a hierarchical cluster analyses of 16S rRNA genes. At a cutoff of 98% sequence identity, one-third of operational taxonomic units (OTUs) with a minimum relative sequence abundance of 0.5% of all bacterial pyrotags occurred at more than one site (Supplementary Figure S2). None of the OTUs occurred at all six tidal sediments sampled along the European Atlantic coastline. To identify potentially habitat-specific ecotypes among the JTB255-MBG, we phylogenetically analyzed nearly full-length 16S rRNA sequences of diverse origin (ARB-Silva SSU Ref release 123, Pruesse et al., 2007). From this analysis, six stable sequence clusters emerged, which comprised mainly sequences of either coastal or deep-sea origin (Supplementary Figure S3). The majority of JTB255-MBG sequences did not show an apparent water depth-dependent phylogenetic pattern. In the future, the integration of full-length 16S rRNA sequences, meta- and single-cell genomes and physico-chemical parameters will be necessary to unravel potential ecotypes and factors shaping the JTB255-MBG communities in marine sediments.
The JTB255-MBG are affiliated with the Woeseiaceae
Given the high sequence diversity and abundance of JTB255-MBG in marine sediments, we aimed at resolving the phylogenetic affiliation of the JTB255-MBG in more detail. To this end, we analyzed high quality, nearly full-length 16S rRNA gene sequences (n=825) using four different treeing methods. In a consensus tree, 96% of all sequences previously classified as JTB255-MBG formed a monophylethic cluster supported by all four treeing methods (Supplementary Figure S3). In contrast to their earlier classification as members of the order Xanthomonadales, our phylogenetic reconstruction indicates that the JTB255-MBG rather forms a stable deep-branching monophyletic cluster within the Gammaproteobacteria. Intriguingly, the recently isolated type strain Woeseia oceani XK5, the only cultured member of the family Woeseiaceae (Du et al., 2016) forms a monophyletic cluster with the JTB255-MBG that is supported by different treeing methods (Supplementary Figure S3). Moreover, the minimum sequence identity within this cluster is 87%, which is above the accepted 86.5% cut-off to discriminate families (Yarza et al., 2014). On basis of these findings, we propose that the JTB255-MBG belongs to the family Woeseiaceae.
To confirm the phylogenetic affiliation of JTB255-MBG with W. oceani, we analyzed a single amplified genome (SAG 1868_B) from a tidal surface sediment in the German Wadden Sea, site Janssand. It encodes 2.27 Mbp and is 48% complete (Table 1). The 16S rRNA displays 93% sequence identity to W. oceani and 92.4% to the gammaproteobacterial metagenomic bin WOR_SG8_31 from White Oak River estuary sediments (Table 1, Baker et al., 2015). From these, we extracted and concatenated 16 ribosomal protein (Rpo) sequences, which are considered to be barely affected by lateral gene transfer (Sorek et al., 2007). Consistent with the 16S rRNA gene phylogeny and supported by four treeing methods, the Rpo-phylogeny confirmed an affiliation of the JTB255-MBG with W. oceani (Supplementary Figure S4). Together, 16S rRNA and Rpo phylogenies provide strong support that the JTB255-MBG are actually members of the family Woeseiaceae. Thus, we henceforth refer to the JTB255-MBG as Woeseiaceae/JTB255.
Genomic repertoire of the Woeseiaceae/JTB255
To recover additional genomic data from this family, we analyzed two incomplete metagenomic bins that we extracted from a bulk metagenome of the Janssand tidal sediment (Table 1, Supplementary Figure S5). The metagenomic bin 20_j1 (2.4 Mbp) was generated by recruitment and assembly of metagenomic reads using SAG 1868_B as reference. Consequently, the bin 20_j1 displays a high average nucleotide identity (ANI) of 99.7% to SAG 1868_B and shows a highly similar genome content. In addition, using GC-content, tetranucleotide frequencies and read coverage, we extracted the metagenomic bin JSS_woes1 (8.1 Mbp). The single-copy genes in this metagenomic bin occur as non-identical dupli- or triplicates, which indicates two to three bacterial genomes in this bin. Two homologous operons encode ribosomal proteins (Rpo) that are most closely affiliated with W. oceani (Supplementary Figure S4, 82 and 84% SI). Consistent with this, the recovered fragments of 5S, 16S, 23S rRNA gene sequences were all highly similar to those of W. oceani (97, 100, 96% SI, respectively). The fact that the metagenomic bin JSS_woes1 attracted in total 6% of all metagenomic reads further supports the high in situ abundance of Woeseiaceae/JTB255 in this tidal sediment.
Potential for chemolithoautotrophy
The recent description of type strain W. oceani XK5 (Du et al., 2016) and the fully sequenced genome (NZ_CP016268) provide first insights into physiology and genetic potential of the Woeseiaceae/JTB255. Physiological tests and the genome content indicate that W. oceani is an obligate chemoorganoheterotroph. This finding, however, contrasts with the recently proposed chemolithoautotrophic potential of the Woeseiaceae/JTB255-MGB (Dyksma et al., 2016). Therefore, we screened SAG 1868_B and all metagenomic bins for pathways that could power growth with inorganic energy and carbon sources.
The (meta)genomes encode three pathways to gain energy from inorganic compounds. SAG 1868_B and bin 20_j1 encode the Sox-pathway (SoxABCDHXYZ) for thiosulfate oxidation (Table 1). For sulfite oxidation, some Woeseiaceae/JTB255 may employ a dissimilatory adenosine-5'-phosphosulfate reductase (AprABM) that is encoded in metagenomic bin JSS_woes1. Moreover, we identified genes encoding an oxygen-tolerant [NiFe] uptake hydrogenase (Hup) plus accessory proteins in SAG 1868_B and in metagenomic bin JSS_woes1 (Table 1).
Furthermore, in metagenomic bin JSS_woes1, we detected genes encoding a Rubisco form II for carbon fixation via the Calvin–Benson-Bassham (CBB) cycle and a phosphoribulokinase (Table 1). These co-localize with three non-CBB-related genes that are most similar to homologs in W. oceani, further supporting an autotrophic potential within the Woeseiaceae (Supplementary Figure S6). To find additional support of a chemolithoautotrophic potential in the Woeseiaceae/JTB255, we re-analyzed the metagenomic bin WOR_SG8_31 (Baker et al., 2015). As the (meta)genomes from the tidal flat, it encodes an aerotolerant [NiFe] uptake hydrogenase, the Sox-pathway for thiosulfate oxidation and a Rubisco for carbon fixation (Table 1).
Collectively, our analyses show that some members of the Woeseiaceae/JTB255 have the genetic potential for chemolithoautotrophy powered by sulfur or hydrogen oxidation. It confirms the previously measured carbon fixation activity of Woeseiaceae/JTB255 cells at our sampling site Janssand (Dyksma et al., 2016).
Potential for heterotrophy
Since the next cultured relative strain W. coeani is an obligate chemoorganoheterotroph, we tested whether the uncultured Woeseiaceae/JTB255 from Janssand tidal sediment also have the potential to use organic molecules as energy (and carbon) sources. The SAG 1868_B and both metagenomic bins encode several organic molecule transporters, alpha-, beta-glucosidases and beta-glucanases, and carbohydrate-active enzymes of glycosyl hydrolase families (Table 1,Supplementary Figure S8). Some also occur in metagenomic bin WOR_SG8_31 and were linked to cellulolysis and amylolysis (Baker et al., 2015). These data indicate a potential to utilize oligo- or polysaccharides, however, we did not detect polysaccharide utilization loci (PUL), typically found in polymer-degrading marine microorganisms (Fernández-Gómez et al., 2013). The type strain W. oceani consumes sugars but does not hydrolyze cellulose, alginate or starch and also seems to lack the typical carbohydrate-hydrolyzing enzymes (Du et al., 2016).
All (meta)genomes encode alcohol dehydrogenases, which is consistent with the observed consumption of glycerol by W. oceani (Du et al., 2016). Moreover, the SAG 1868_B, metagenomic bins and W. oceani encode diverse proteases and peptidases (Supplementary Figure S8). For example, dipeptidyl-peptidases M48 and S46 occur in SAG 1868_B but not in any autotrophic gammaproteobacterial sulfur-oxidizer cultured to date. Dioxygenases, such as tryptophan-dioxygenase are found in all tested (meta)genomes and are employed to aerobically cleave aromatic ring structures (Supplementary Figure S8). Consistent with this, W. oceani was isolated on peptone agar and displays proteolytic enzyme activities (Du et al., 2016). In summary, the ability to consume organic compounds such as carbohydrates and peptides appears to be common among the studied (meta)genomes of Woeseiaceae/JTB255. The varying peptidase/GH ratios (Table 1), however, also suggest distinct substrate preferences among the studied members of Woeseiaceae/JTB255.
Aerobic and anaerobic respiration
Our (meta)genomic analyses and the physiological tests with W. oceani (Du et al., 2016) indicated the capability for aerobic and anaerobic growth. Oxygen is respired using aa3-type and cbb3-type cytochrome c oxidases. For oxygen-independent respiration SAG 1868_B, metagenomic bins JSS_woes1 and WOR_SG8_31, and W. oceani may employ consistently a truncated denitrification pathway, including the periplasmic dissimilatory nitrite NirS and the membrane-bound nitric oxide reductase NorB (Supplementary Figure S8). While W. oceani does not respire nitrate (Du et al., 2016) and only possesses nirS and norB genes, we cannot exclude that we missed additional denitrification genes in the non-sequenced parts of the (meta)genomes. However, truncated denitrification pathways and modularity are common among denitrifying microorganisms (Graf et al., 2014) and also appear to drive denitrification at our sampling site (Marchant et al., under review). In fact, coastal sandy sediments have a more important role in N-loss than assumed and may account for a significant fraction of the global oceanic emission of the potent greenhouse gas nitrous oxide (Marchant et al., under review). If a truncated denitrification is common among the Woeseiaceae/JTB255, this ubiquitous and abundant group might substantially contribute to nitrous oxide emissions from coastal sediments.
Conclusion
Our study highlights the role of the Woeseiaceae/JTB255 in marine surface sediments. The genomic repertoire presented in this study and the physiology of the first isolated strain W. oceani suggest that this family is heterogeneous and covers a broad physiological spectrum ranging from facultative sulfur- and hydrogen-based chemolithoautotrophy to obligate chemorganoheterotrophy. This could provide adaptations to various biogeochemical settings and possibly explains their success in marine sediments worldwide. Given their substantial sequence and cell frequencies in sediments across the oceans, we propose that the Woeseiaceae/JTB255 are important members of microbial benthic communities and are likely among the most abundant microorganisms in both deep-sea and coastal sediments. Thus, it is imperative to unravel their in situ activity and function in carbon, sulfur and nitrogen cycling in diverse types of marine sediment.
References
Amaral-Zettler L, Artigas LF, Baross J, Bharathi PAL, Boetius A, Chandramohan D et al. (2010). A global census of marine microbes. In: Life in the World’s Oceans. Wiley-Blackwell, pp 221–245.
Baker BJ, Lazar CS, Teske AP, Dick GJ . (2015). Genomic resolution of linkages in carbon, nitrogen, and sulfur cycling among widespread estuary sediment bacteria. Microbiome 3: 14.
Bienhold C, Zinger L, Boetius A, Ramette A . (2016). Diversity and biogeography of bathyal and abyssal seafloor bacteria. PloS One 11: e0148016.
Bowman JP, McCammon SA, Dann AL . (2005). Biogeographic and quantitative analyses of abundant uncultivated γ-proteobacterial clades from marine sediment. Microb Ecol 49: 451–460.
Burdige DJ . (2007). Preservation of organic matter in marine sediments: controls, mechanisms, and an imbalance in sediment organic carbon budgets? Chem Rev 107: 467–485.
Du Z-J, Wang Z-J, Zhao J-X, Chen G-J . (2016). Woeseia oceani gen. nov., sp. nov., a chemoheterotrophic member of the order Chromatiales, and proposal of Woeseiaceae fam. nov. Int J Syst Evol Microbiol 66: 107–112.
Dyksma S, Bischof K, Fuchs BM, Hoffmann K, Meier D, Meyerdierks A et al. (2016). Ubiquitous Gammaproteobacteria dominate dark carbon fixation in coastal sediments. ISME J 10: 1939–1953.
Fernández-Gómez B, Richter M, Schüler M, Pinhassi J, Acinas SG, González JM et al. (2013). Ecology of marine Bacteroidetes: a comparative genomics approach. ISME J 7: 1026–1037.
Graf DR, Jones CM, Hallin S . (2014). Intergenomic comparisons highlight modularity of the denitrification pathway and underpin the importance of community structure for N2O emissions. PloS One 9: e114118.
Liu J, Liu X, Wang M, Qiao Y, Zheng Y, Zhang X-H . (2015). Bacterial and archaeal communities in sediments of the North Chinese marginal seas. Microb Ecol 70: 105–117.
Marchant HK, Tegetmayer HE, Ahmerkamp S, Holtappels M, Lavik G, Graf J et al. Metabolic specialization of denitrifiers in permeable sediments leads to N2O emissions (under review).
Orcutt BN, Sylvan JB, Knab NJ, Edwards KJ . (2011). Microbial ecology of the dark ocean above, at, and below the seafloor. Microbiol Mol Biol Rev 75: 361–422.
Pruesse E, Quast E, Knittel K, Fuchs BM, Ludwig W, Peplies J et al. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35: 7188–7196.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41: D590–D596.
Ruff SE, Probandt D, Zinkann AC, Iversen MH, Klaas C, Würzberg L et al. (2014). Indications for algae-degrading benthic microbial communities in deep-sea sediments along the Antarctic Polar Front. Deep Sea Res PT II 108: 6–16.
Schauer R, Bienhold C, Ramette A, Harder J . (2010). Bacterial diversity and biogeography in deep-sea surface sediments of the South Atlantic Ocean. ISME J 4: 159–170.
Sorek R, Zhu Y, Creevey CJ, Francino MP, Bork P, Rubin EM . (2007). Genome-wide experimental determination of barriers to horizontal gene transfer. Science 318: 1449–1452.
Tully BJ, Heidelberg JF . (2013). Microbial communities associated with ferromanganese nodules and the surrounding sediments. Front Microbiol 4: 161.
Yarza P, Yilmaz P, Pruesse E, Gloeckner FO, Ludwig W, Schleifer K-H et al. (2014). Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol 12: 635–645.
Zinger L, Amaral-Zettler LA, Fuhrman JA, Horner-Devine MC, Huse SM, Welch DBM et al. (2011). Global patterns of bacterial beta-diversity in seafloor and seawater ecosystems. PLoS One 6: e24570.
Acknowledgements
We greatly acknowledge the excellent service by Dr Claudia Bergin from the Microbial Single Cell Genomics facility at SciLifeLab in Uppsala, Sweden. Kenneth Wasmund provided helpful comments on the manuscript. We thank Professor Rudolf Amann at MPI Bremen/Germany and Professor Michael Wagner at DoME Vienna/Austria for the excellent support. Jörg Wulf and Christian Castro-Romero provided great technical assistance. The Max Planck Society, Germany, funded this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Mußmann, M., Pjevac, P., Krüger, K. et al. Genomic repertoire of the Woeseiaceae/JTB255, cosmopolitan and abundant core members of microbial communities in marine sediments. ISME J 11, 1276–1281 (2017). https://doi.org/10.1038/ismej.2016.185
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2016.185
This article is cited by
-
A first report on prokaryotic diversity in northwestern Arafura deep-sea sediments, Indonesia
Scientific Reports (2024)
-
Genomic insights into cryptic cycles of microbial hydrocarbon production and degradation in contiguous freshwater and marine microbiomes
Microbiome (2023)
-
Genome-resolved metagenomics of Venice Lagoon surface sediment bacteria reveals high biosynthetic potential and metabolic plasticity as successful strategies in an impacted environment
Marine Life Science & Technology (2023)
-
Polychaete Bioturbation Alters the Taxonomic Structure, Co-occurrence Network, and Functional Groups of Bacterial Communities in the Intertidal Flat
Microbial Ecology (2023)
-
Microbial diversity and functional profiling in coastal tidal flat sediment with pollution of nutrients and potentially toxic elements
Journal of Soils and Sediments (2023)
