
Abstract
To explain differences in gut microbial communities we must determine how processes regulating microbial community assembly (colonization, persistence) differ among hosts and affect microbiota composition. We surveyed the gut microbiota of threespine stickleback (Gasterosteus aculeatus) from 10 geographically clustered populations and sequenced environmental samples to track potential colonizing microbes and quantify the effects of host environment and genotype. Gut microbiota composition and diversity varied among populations. These among-population differences were associated with multiple covarying ecological variables: habitat type (lake, stream, estuary), lake geomorphology and food- (but not water-) associated microbiota. Fish genotype also covaried with gut microbiota composition; more genetically divergent populations exhibited more divergent gut microbiota. Our results suggest that population level differences in stickleback gut microbiota may depend more on internal sorting processes (host genotype) than on colonization processes (transient environmental effects).
Similar content being viewed by others

Introduction
Metacommunity theory is a branch of ecology describing the dynamics and composition of patchily distributed species assemblages (Levins, 1970). The microbiota of the vertebrate gastrointestinal tract provides an excellent example of a metacommunity (Costello et al., 2012; O'Dwyer et al., 2012), comprising diverse species assemblages distributed across many discrete and temporary patches (individual hosts). In metacommunities, the species composition within any single patch (host) depends on two processes. First, colonization by microbes from the external environment continually adds species to a local community. Second, whether these colonists persist or are lost depends on ecological interactions within the patches (for example, among microbes, or between the host and microbiota), which ecologists call ‘filtering’ or within-patch dynamics. The mechanisms governing colonization and filtering of gut microbiota remain poorly understood, yet this knowledge is vital for treating diseases associated with dysbiosis (Hofer and Speck, 2009; Carlisle et al., 2011; Frazier et al., 2011; Haiser and Turnbaugh, 2012; Wong et al., 2012; Atarashi et al., 2013). We studied wild populations of threespine stickleback (Gasterosteus aculeatus), a small fish with variable ecology, to identify major pathways of microbial colonization (via water- or food-borne microbes). We also asked to what extent do population differences in microbiota reflect differential colonization versus differences in host genotype (which likely represents host-specific filtering dynamics)?
Microbial colonization of the vertebrate gut begins at birth through contact with parents and the external environment (Bergh, 1995; Hansen and Olafsen, 1999; Dominguez-Bello et al., 2010; Pantoja-Feliciano et al., 2013). Colonization continues throughout life by microbes from ingested materials, such as food and liquid, or via contact with microbes on other organisms or abiotic surfaces (Liston, 1956; Campbell and Buswell, 1983; Meadow et al., 2013). Most of these colonists are transient, either passing through the gut or dying. Nevertheless, host populations exposed to different microbial colonists may exhibit divergent microbiota. For example, because water salinity affects ambient microbial taxa, fish gut microbiota communities differ between fresh and marine populations (Sullam et al., 2012). However, the sources of microbial colonists and their relative importance remain unknown for many organisms. Here, we quantify the contribution of two major sources of fish gut colonists: microbes from invertebrate prey, and in the ambient water.
Once colonizing bacteria enter the host’s gut, the colonists may decline in abundance (if they are excreted or reproduce poorly), or may successfully establish a self-perpetuating local population. Whether a colonizing species becomes a member of the community depends on the relative rates of recurrent colonization versus local extinction (for example, the balance of colonization versus filtering dynamics). The filtering process, in metacommunity theory, can depend on several factors. First, arriving microbes must have an appropriate spatial and nutritional niche within the host gut (Laparra and Sanz, 2010; Faith et al., 2011; Nicholson et al., 2012). In particular, host food composition affects the persistence of extant microbes through nutritional inputs needed to sustain microbial reproduction (Hildebrandt et al., 2009; Wu et al., 2011; Parks et al., 2013). Second, food intake and digestion rates affect the speed of food passage through the gut and thus the rate at which microbes are lost via excretion. Third, colonist persistence depends on interactions with other microbes (Lozupone et al., 2012), including resource competition (Mahowald et al., 2009), allelopathy (Leão et al., 2012) and cross-feeding (Rey et al., 2010). Fourth, colonizing microbes must survive potential host immune responses (Gomez and Balcazar, 2008; Maslowski and Mackay, 2011; Round et al., 2011; Carvaho et al., 2012; Hooper et al., 2012; Biswas and Kobayashi, 2013; Cebula et al., 2013; Robertson and Girardin, 2013; Bolnick et al., 2014b). Genetic diversity within and among host populations is known to generate variation in immune response, which may in turn contribute to filtering of microbial colonists. Here, we show that within- and among-population differences in gut microbiota reflect host genetic diversity, even after accounting for differential colonization. We use these findings to infer that metacommunity structure of the stickleback microbiota is more strongly driven by host genotype (likely reflecting species filtering processes (Ley et al., 2008; Benson et al., 2010; Kovacs et al., 2011)) than by colonization.
Materials and methods
Study system
The threespine stickleback is a widely used model organism in evolutionary biology because of its repeated post-glacial colonization of freshwater habitats from marine populations, leading to replicate convergent evolution in different populations (Reusch et al., 2001). Recently established freshwater populations diverged from their marine ancestors (Jones et al., 2012), and also exhibit adaptive divergence among populations in disparate freshwater habitats (small versus large lakes, or streams) (Lavin and McPhail, 1985; Hendry et al., 2009, 2013). These habitats vary in abiotic variables such as water chemistry and temperature, and in biotic features such as prey availability, both of which may separately affect microbial colonization of the host gut. Stickleback populations also exhibit varying levels of genetic divergence, arising from both neutral evolution and divergent natural selection. This genetic divergence is especially striking for some immune genes, including but not limited to MHC class II (Stutz and Bolnick, 2014), which can regulate microbial persistence within the host gut (Bolnick et al., 2014a). Consequently, environmental and host genetic divergence among stickleback populations provides an opportunity to partition the effects of colonization and host genetic filtering on gut microbial metapopulation structure in a wild animal species.
We compared the gut microbiota composition and diversity of 10 stickleback populations, including two marine and eight freshwater populations. The freshwater populations come from a single watershed (all <7 km apart). The two populations of anadromous marine stickleback were from the closest estuaries immediately north and south of this drainage (72 km apart). By sampling freshwater sites within a few kilometers of each other, we minimize environmental heterogeneity. Water temperatures at these sites are typically within a few degrees of each other, and conductivity, pH and dissolved oxygen levels are highly similar. The ecological differences that do exist between our sample sites (particularly between lake and stream habitats, and between large and small lakes) are largely unrelated to genetic differentiation between sites, which primarily reflects spatial connectivity (Caldera and Bolnick, 2008). This facilitates our goal of separately estimating effects of populations’ environmental and genetic variation in gut microbiota. A further benefit is that all freshwater populations share a common ancestor since deglaciation 12 000 years ago (Hagen and McPhail, 1970; Caldera and Bolnick, 2008), providing a known time frame for gut microbial divergence.
Using these samples, we asked whether the gut microbiota differs among geographically and genetically related populations. We then tested whether population-level differences can be attributed to differential colonization by water- or food-borne microbes, or attributed to filtering based on host genotype. We use these results to draw inferences regarding the relative role of colonization and filtering dynamics in a wild gut microbial metapopulation.
Sample acquisition
From 20 June to 11 July, 2012, we collected 182 stickleback from 10 sites (Supplementary Tables S1 and S2) including six lakes, two streams and two estuaries on Vancouver Island, British Columbia (Figure 1). Stickleback were trapped in unbaited minnow traps along the shoreline of each site, at depths ranging from 0.5 to 3.0 m and typically 1–5 m offshore.

Map of Vancouver Island, British Columbia, Canada (lower left box), showing the location of two estuary sample sites, and the Amor de Cosmos watershed. The detailed inset map (upper right) shows the six lake and two stream sites sampled from within the Amor de Cosmos watershed.
We also sampled typical stickleback prey: from each site at least 20 macroinvertebrates were collected by scooping mud with a small aquarium net, and four zooplankton samples were collected by towing a 50 μm Wisconsin-style plankton tow just below the water surface for 20–50 m. Examining these samples under a dissecting microscope, we retained individual invertebrates that are typically consumed by stickleback in a given lake (Snowberg et al. (in press)). Note, however, that we were unable to sample all possible invertebrate prey from a given lake, both because of limited sampling per site and because some prey species are only available at other times of year or certain times of day (all samples were collected between 1600 and 2000 hours). Finally, four water samples (40 ml) were collected just below the surface (5 cm) to identify ambient microbes at each site. All samples were immediately frozen. For each type of sample (fish, prey, water), we collected from various locations within each sample site to maximize surface coverage and to maximize representation of the site’s communities. However, it was not our goal, nor was it feasible, to comprehensively sample environmental and invertebrate-associated microbiota, which may vary over diel or seasonal cycles, as well as potentially varying between innumerable microhabitats within a given location. It is therefore possible that some differences between fish gut microbiota and environmental microbiota are due to necessarily incomplete sampling of both types of microbial communities.
In the laboratory, we partially thawed stickleback and dissected (using sterile tools and work surface) each fish to extract the whole intestine (~35 mg) for microbial DNA extraction. In many species the gut microbiota varies along the length of the digestive tract (Dethlefsen et al., 2006); we averaged across such variation by using the entire intestine because the spatial structure of stickleback gut microbiota is unknown. Also, by sampling the entire intestine we include bacteria in the gut lumen, as well as those associated with mucosal surfaces. The latter are more likely to remain long-term residents. Lumen microbiota are more likely to be recent colonists imported with prey items, and are more likely to be rapidly excreted in feces. However, both lumen- and mucosal-associated bacteria may be important members of the microbial community from the standpoint of digestion, host nutrition and immunity.
We determined the sex of each fish via gonad morphology. Macroinvertebrates and zooplankton were sorted by taxon (Supplementary Table S1). DNA was immediately isolated from separate individuals for larger invertebrates, or from pools combining a few conspecific individuals for smaller insect larvae, or pools of up to 100 individuals for small zooplankton taxa (Supplementary Table S3).
Amplification, sequencing and analysis of 16S rRNA genes
We extracted DNA from stickleback, macroinvertebrates and zooplankton using Powersoil DNA Isolation Kits (MoBio Laboratories, Carlsbad, CA). The Powersoil protocol was modified as described in Bolnick et al. (2014c). After filtering water samples we extracted DNA following the protocol of the MoBio PowerWater DNA isolation kit.
We amplified and sequenced the V4-V5 hypervariable regions of the 16S rRNA (positions 515 to 806, based on E. coli numbering) gene in all samples using the procedure in Bolnick et al. (2014c) except for the following modifications. The forward primers contained the 5′ Illumina sequencing adapter, a 10 nt pad sequence, followed by the 515 16S specific linker and primer sequence. The reverse primer contained the 3′ reverse complement of the Illumina sequencing adapter, the 12 nt Golay barcode, a 10 nt pad sequence followed by the 16S specific 806R reverse linker and primer. Sequencing was done on an Illumina MiSeq genome sequencer at the University of Texas at Austin.
Data analysis was performed using QIIME v1.7 (Caporaso et al., 2010). Initial quality filtering was done as in (Bokulich et al., 2013; Supplementary Table S4). To focus the analysis on archaeal and bacterial taxa, OTUs (operational taxonomic units) were picked using a closed-reference OTU picking protocol against the Greengenes database (DeSantis et al., 2006; available at http://greengenes.lbl.gov/Download/Sequence_Data/Fasta_data_files/Caporaso_Reference_OTUs/gg_otus_4feb2011.tgz), prefiltered at 97% identity: between 40 and 70% of reads were discarded. As described in Bolnick et al. (2014b), the vast majority of our discarded reads were either host mitochondrial sequence or phiX, which was spiked into the library to increase read diversity. Some discarded sequences had no blast hits at all and likely represent sequencing error. As an additional quality-filtering step, OTUs represented by <10 sequences were discarded, as described in Caporaso et al. (2011), to focus on high-quality sequences. Closed-reference OTU picking of our stickleback microbiota sequences retained most (~75%) of the sequences obtained by open-reference OTU picking (which we did for comparison). Open and closed-reference OTU picking yields highly correlated alpha and beta diversity measures in stickleback. We focused on closed-reference OTU picking for these analyses because this method yields higher-quality taxonomic identifications and a more reliable OTU phylogenetic tree (which is based on the full-length sequences rather than on short tags) than do open-reference methods.
Taxonomic assignments for OTUs were based on the Greengenes reference sequence defining that OTU, and the Greengenes tree was used for computing phylogenetic diversity metrics. Because diversity metrics are sensitive to sampling effort, we rarefied the data to 3000 sequences per sample; diversity measures are highly correlated across various levels of rarefaction in the stickleback gut (Bolnick et al., 2014b), and using the lower rarefaction level allows us to retain more samples. Alpha diversity of individual samples was calculated post-rarefaction as phylogenetic diversity, Chao1 and species richness. Between-sample, pairwise diversity was calculated with unweighted or weighted UniFrac (weighted for OTU relative abundance) (Lozupone and Knight, 2005). We then performed principal coordinate analysis (PCoA) on the resulting distance matrices for lake fish only.
Statistical analysis
We used multiple measures of microbial variation (UniFrac distances, PCoA scores or alpha diversity metrics) in several statistical tests. First, we used PERMANOVAs to test for microbial divergence among populations. Second, we used several complimentary approaches (Mantel tests, t-tests of UNIFRAC distances, and Sourcetracker) to test whether geographic variation in water or prey microbiota explains variation in host microbiota. Third, we used previously published population genetic data from the same populations examined here, to estimate pairwise genetic divergence between populations (Rst) and mean heterozygosity within each population. We then tested whether host genetic variation (within- and between populations) is correlated with within- and between-population differences in gut microbiota composition, controlling for geographic variation and environmental microbiota. Additional details of each test are given alongside the corresponding results, below, for clarity. For each of the statistical analyses involving UniFrac distances or PCoA scores we carried out two separate tests: using either unweighted or weighted Unifrac distances (or PCoA scores calculated from UniFrac distances). The following analyses were carried out in R (R Development Core Team, 2012), unless specified otherwise.
Results
Microbial divergence among populations
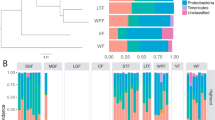
We first tested whether gut microbiota composition differed among all 10 stickleback populations. We used a nested PERMANOVA (10 000 permutations) to estimate the roles of habitat (lakes versus streams versus estuaries) and population (nested within habitat) in explaining variation in UniFrac distances between individual fish. Substantial gut microbiota differentiation exists among populations (unweighted UniFrac P<0.001, F=3.569; weighted UniFrac P<0.001, F=3.266), and somewhat between habitats (unweighted F=2.228, P=0.049; weighted F=1.773, P=0.089; Figures 2a and 3a). Using a regression of divergence time estimates (from Caldera and Bolnick, 2008) for three population pairs (2000 to 5000 years diverged), we quantified the rate of gut microbiota divergence (UniFrac distance between populations/mean UniFrac distance within those populations) to be increasing 0.029 unweighted UniFrac distances (0.096 weighted) every 1000 years.

(a) By habitat type, the first two axes from a Linear Discriminant Analysis (LDA) describing variation in the first 21 unweighted axes (the number of axes that individually describe at least 1% of variation) from the fish-only PCoA. (b) By population, the first two axes from an LDA describing variation in the first 27 unweighted axes from the lake fish only PCoA.
Next, we focused exclusively on the six lake populations. We used a PERMANOVA (10 000 permutations) to test whether gut microbiota composition varied among populations or between sexes, retaining R2 values as a measure of effect size for each model term. The six lake populations differed from one another using unweighted and weighted UniFrac distances (both P<0.001; Figure 2b). Among lakes, population explained 19.5% of unweighted UniFrac variation (weighted UniFrac, 15.3%). However, sex did not have a significant main effect (unweighted UniFrac R2=0.012, P=0.150; weighted R2=0.004, P=0.550) or interaction with population (unweighted UniFrac R2=0.051, P=0.142; weighted R2=0.066, P=0.095). We identified specific microbial taxa important in distinguishing among habitat types and among populations using QIIME’s supervised learning functionality (Knights et al., 2011) (Supplementary Figures S1 and S2; Supplementary Tables S5 and S6).
Colonization by environmental microbiota
We next quantified the contributions of water- and food-associated microbiota variation in the stickleback gut microbiota composition. Ingested water and food import recurrent microbial colonists of the stickleback gut. These colonizing microbes may (i) be transient and rapidly excreted, (ii) be moribund because they are unable to survive or reproduce in the vertebrate gut or (iii) become permanent residents of the stickleback gut. Note that, consistent with metacommunity theory, we use the term colonization to refer to newly arrived species without presupposing a long-term outcome (persistence or elimination).
Considering all stickleback samples together, a comparable number of gut microbe OTUs were shared with water (38.0%) and invertebrate (43.0%) microbial communities (Figure 3b). There was also overlap between water and invertebrate communities. Note that these counts of shared microbial OTUs are sensitive to sampling effort; more extensive sampling of water and invertebrate microbiota would presumably reveal additional microbes and might therefore increase these estimates of overlap with the fish gut microbiota.

(a) Number of OTUs shared among stickleback from different habitats after rarefaction to 3000 sequences per individual. (b) Number of OTUs shared among stickleback, invertebrates and water microbiotas after rarefaction to 3000.
We then used Bayesian community-level source tracking (Knights et al., 2011) to estimate how much of the stickleback gut microbiota is from water, or invertebrate prey sources (after filtering OTUs found in fewer than 1% of all samples): on average 12.6% of the fish gut microbiota was from water sources, 73.3% from prey sources and 14.1% unknown (<0.05% of all samples came from presumed human gut, oral, or skin contamination). This analysis does not consider all source-sink scenarios (for example, fish-fish transmission or fish-to-water), nor does our study consider other potential environmental sources (for example, sediment, vegetation, allochthonous debris) or all possible prey. However, our data suggest that the gut microbiota is assembled partly via colonization by both water- and food-associated microbes, with food being the more substantial of the two sources. A caveat is that we cannot rule out the possibility that water microbes are important colonists but are disproportionately excluded by the host immune system or gut environment.
A follow-up question is whether the among-population gut microbiota differences we observe are due to differential exposure to environmental microbes. First, we focus on the overall resemblance between individual fish and the microbiota of the local water. Water microbial communities differed among sample sites (unweighted UniFrac PERMANOVA P<0.001, R2=0.739; weighted UniFrac P<0.001, R2=0.909). If fish gut microbiota are predominantly assembled via water-associated colonists, then we expect fish gut microbiota to most closely resemble the water microbiota from their own site, compared with other sites’ water microbiota. To test this prediction, we did a one-tailed t-test using pairwise UniFrac distances between individual stickleback and individual water samples. We did not find higher resemblance between stickleback gut microbiota and their local water versus foreign water microbiota (unweighted UniFrac t=0.497, P=0.685; weighted t=−0.596, P=0.281).
Using SourceTracker to estimate the contribution of native water versus foreign water to stickleback microbiota, we found no evidence that individual stickleback are colonized by local water microbes more strongly than by foreign water microbes (one-tailed t=0.610, P=0.278). Similarly, stickleback OTUs were not shared more often with water samples from their native water versus from foreign water (one-tailed t=0.593, P=0.283).
Finally, we can focus on the aggregate microbial metacommunity in each host population. We evaluated whether the matrix of between-population gut microbial distances (mean UniFrac distances) is correlated with the matrix of between-population water microbial distances. Geographic distance was correlated with mean pairwise UniFrac distances of both water (unweighted UniFrac R=0.691, P=0.012; weighted R=0.561, P=0.017) and gut microbiota (unweighted Unifrac R=0.656, P=0.010; weighted R=0.170, P=0.264) among the 10 sample sites. Controlling for geographic distance as a confounding variable using a partial Mantel test, we found no correlation between gut microbiota distance and water microbiota distance (unweighted UniFrac R=0.176, P=0.210; weighted R=0.076, P=0.333). The effect of geography is driven by the fact that the marine sites are more distant, and more environmentally distinct, from all other sites (which are comparatively tightly clustered). In conclusion, although water microbes are an important source of colonists for the fish gut (as shown above), multiple analyses indicate that geographic differences in water microbes are not responsible for the geographic differences in fish gut microbiota. Note that we do not rule out associations between microbe OTUs that are rare in both water and stickleback. However, these rare OTUs would contribute little to overall microbial variation here.
Exposure to prey microbiota
Microbial differences between fish populations might also arise from colonization by different food-associated microbes. Diet can affect exposure because commonly eaten prey carry different microbes in different lakes, because fish from different lakes eat different kinds of prey (Matthews et al., 2010), or because the nutrients in different prey items favor the growth of different microbes in the fish gut (Bolnick et al., 2014b). These mechanisms are not mutually exclusive, so for present purposes we lump them together and ask whether fish gut microbiota resemble the microbiota of the dominant prey in their habitat more than microbiota of dominant prey in other habitats. Dominant prey within a location were those that were both particularly abundant in our environmental samples, and thus commonly found in stickleback stomach contents (Snowberg et al. (in press)). We used approaches similar to the tests of water-microbe effects, described above, to compare fish microbiota with local versus foreign prey microbes.
Pairwise unweighted UniFrac distances between fish gut microbiota and local versus foreign invertebrates (not necessarily the same species across sample sites) demonstrated that fish gut microbiota disproportionately resembled the microbiota of local, rather than foreign, invertebrate prey (t=−2.156, P=0.024; not significant with weighted UniFrac: t=−1.336, P=0.101). SourceTracker scores assigning fish microbiota to local versus foreign invertebrates revealed a marginally significant tendency for fish gut microbiota to come from local rather than foreign prey (t=1.774, P=0.053). However, the proportion of OTUs shared between fish and invertebrate samples (like above) did not reveal a convincing bias towards local sources (t=0.912, P=0.189). Finally, lakes with more similar invertebrate microbiotas also had more similar gut microbiotas using unweighted UniFrac (Mantel R=0.395, P=0.029; but not weighted: R=−0.234, P=0.856), while controlling for geographic distance. Thus, geographic variation in the microbiota of stickleback prey (mostly insect larvae and crustaceans) is associated with the geographic variation in stickleback microbiota.
Genetic versus microbial divergence
We used previously published microsatellite data (Caldera and Bolnick, 2008) from the six lakes studied here to test whether more genetically divergent populations are more microbially divergent. We used estimates of population genetic divergence (Rst) between each pair of lakes in partial Mantel tests to determine whether between-population gut microbial distance (mean UniFrac) is correlated with between-population genetic and geographic distances. Rst and mean UniFrac distance were positively correlated among lakes while controlling for geographic distances (unweighted UniFrac Mantel R=0.651, P=0.020; weighted R=0.511, P=0.038, Figure 4). Microbiota-Rst correlations were significant with or without geographic distance as a covariate.

Linear regression of genetic divergence (Rst, based on microsatellite allele frequencies reported in Caldera and Bolnick 2008) among six lake populations versus mean microbial phylogenetic distance (mean unweighted UniFrac). Mantel R=0.651, P=0.020.
Among-lake geographic distance was not correlated with genetic differentiation (R=0.012, P=0.468) or gut microbiota distance (unweighted UniFrac R=0.206, P=0.250; weighted R=0.017, P=0.461). The lack of geographic distance effect, when analysing just lakes, is different from the strong effect of distance seen when we included marine sites (described above). This is because marine sites are both far more distant from the lakes and streams, and more ecologically and microbially divergent. Omitting these far-distant marine sites thus eliminates the effect of geographic distance on microbial differences between populations.
Stickleback populations are known to diverge in diet composition (Matthews et al., 2010; Snowberg et al. (in press)), which raises the possibility that more genetically divergent host populations might be exposed to more divergent environmental microbes. To control for this possibility, we removed environment-associated OTUs from the data set and re-analyzed the association between genetic and microbial divergence among populations. Removing water- or invertebrate-associated microbes, or both, increases the strength and statistical significance of the positive association between host genetic distance and gut microbiota divergence (Supplementary Table S7).
Finally, we used a multivariate analysis of variance (MANOVA) to test whether population differences in gut microbiota composition (the first three unweighted or weighted PCoA scores) covary with genetic diversity within populations. We measured genetic diversity as the mean within-individual heterozygosity, at the six microsatellite loci from Caldera and Bolnick. (2008). This yields an estimate, for each lake, of genetic diversity and thus effective population size. We found that gut microbiota composition covaried with within-population genetic diversity using unweighted PCoA (Pillai=0.971, P=0.043) but not weighted (Pillai=0.394, P=0.753). Note that populations, not individual fish, constitute the level of replication in this analysis, and therefore it is not necessary that estimates of population heterozygosity and population microbiota composition come from the same individual animals. Levels of heterozygosity and population divergence, with these neutral markers, are quite stable from year to year in stickleback from this region.
Microbial alpha and beta diversity within populations
We next examined population-level differences in microbial community diversity. We used ANOVAs comparing within-fish diversity (alpha diversity; measured as phylogenetic diversity, Chao1 and species richness) among populations, habitats and sex. Alpha diversity differed among populations for all metrics (each P<0.001; Figure 5).

Alpha diversity levels (# OTUs) of stickleback gut microbiotas after rarefaction to 3000. Metrics phylogenetic diversity, chao1 and species richness all varied among populations: each P<0.001, and F=4.667, 4.452, 5.497, respectively. White=estuary, light gray=stream, dark gray=lake.
Similarly, we used an ANOVA to compare between-fish diversity (beta diversity: mean pairwise UniFrac distances among all individuals in a population) among populations. Because this test entails pairwise comparisons among individuals, we used a Monte Carlo permutation approach to obtain P-values for this ANOVA, shuffling fish among populations and calculating resulting null F statistics; repeating this permutation 1000 times provided a null distribution against which we tested the observed F statistic. Between-fish diversity also differed among populations (P=0.002 and P=0.001 for unweighted and weighted UniFrac, respectively).
Larger and deeper lakes tend to have higher effective population sizes and more genetic heterozygosity (Caldera and Bolnick, 2008), so we next tested for associations between microbial diversity and heterozygosity within populations. There was no significant relationship between mean heterozygosity and mean microbial alpha diversity (for any metric, all P>0.1). However, more heterozygous populations tended to exhibit less beta diversity (among-individual variation) in their gut microbiota (weighted UniFrac R2=0.769, P=0.014; unweighted R2=0.446, P=0.088).
Environmental covariates
We tested whether lake geomorphology (mean depth) covaries with fish gut microbiota diversity. Geomorphology data were acquired from Caldera and Bolnick (2008). Previous studies have found that, within a single lake population, individual stickleback differ in prey preferences (Matthews et al., 2010) and exhibit corresponding differences in gut microbiota composition (Bolnick et al., 2014c). Very large and very small lakes tend to be dominated by limnetic- and benthic-specialists, respectively, while intermediate-sized lakes harbor a mixture of both and thus have greater among-individual diet variation (Bolnick, 2011). If among individual diet variation confers greater among individual microbial variation, we expected a negative quadratic (-shaped) relationship between lake size and microbial beta diversity. Quite the contrary, quadratic regression of beta diversity on lake size (depth) revealed a significant positive quadratic relationship (unweighted UniFrac distances R2=0.818, P=0.028; weighted R2=0.467, P=0.167; Figure 6). This indicates that among-individual microbial variation is actually greater in small shallow and larger deeper lakes, where among-individual diet variation is typically reduced. Microbial beta diversity is lower in mid-sized lakes where diet heterogeneity is typically expected. We emphasize that this result has low power, relying on only six lakes. However, this represents one of the first tests of whether abiotic (geomorphological) variables affect gut microbial composition and diversity.

Regression of mean lake depth2 with mean between-fish unweighted UniFrac distance among the six lake populations, plus upper and lower confidence intervals.
Discussion
We found that threespine stickleback populations differ in gut microbial composition, within-host diversity and between-host diversity. This among-population variation was attributable both to general habitat type (lake, stream or estuary) and to differences among populations of the same habitat type (lakes). Population effects were greater than habitat effects in explaining microbial variation, and were far more important than variation due to host sex.
After accounting for habitat and population effects, there remained substantial residual variation in microbial community composition, which likely reflects among-host variation in diet (Bolnick et al. 2014b, 2014c) and genotype (Bolnick et al., 2014a). We conclude that there is significant variation in stickleback gut microbiota across multiple nested levels: microbial diversity within individual fish hosts, microbial variation among co-occurring host individuals and microbial variation among host populations. The next challenge is to evaluate the extent to which this variation is attributable to differences in colonization and/or host genotype.
Differential colonization
Our data demonstrate that population differences in gut microbiota composition can be attributed partly to differential colonization by microbes from the external environment. Variation in habitat type (lake, stream or estuary) can cause differential exposure to microbes. Numerous biotic and abiotic variables distinguish these habitat types and potentially influence the gut microbiota. For example, salinity is the predominant variable distinguishing environmental microbes worldwide (Lozupone and Knight, 2007) and likely causes differences in the microbial communities between our freshwater and marine sites. Correspondingly, several of the most important OTUs distinguishing among habitat types were marine cyanobacteria (Prochlorococcus marinus). However, our analyses did not find evidence that water-microbiota differences influence differences in the gut microbiota among populations. This is surprising, given that water-derived microbes contribute an estimated 12.6% of the stickleback gut microbiota, and water samples are distinct among sites.
Previous research in a wide variety of species (including stickleback) has shown that individual diet influences gut microbial composition (Turnbaugh et al., 2008; Muegge et al., 2011; Wu et al., 2011; Bolnick et al., 2014b, 2014c). Such diet effects, however, can arise either from differential colonization by food-associated microbes or nutritional effects on microbial persistence. We found evidence for the former (colonization effects), because among-lake differences in the microbiota of commonly eaten prey were associated with among-lake differences in fish gut microbiota. That is, according to some metrics we examined, stickleback gut microbes disproportionately resembled the microbiota of invertebrates from fishes’ native lake, as opposed to microbiota of invertebrates from foreign lakes. Thus, stickleback gut microbiota tends to be more similar to food-associated than water-associated microbes, and geographic variation in food microbiota contributes more to geographic variation in fish microbiota. The food-microbes involved in this relationship may be more long-term inhabitants of the gut, or more transient inhabitants. Additional work will be needed to test whether this overlap arises from shared symbionts, or whether fish guts contain transient microbes imported with food but unable to persist within the gut. However, even transient microbes might contribute to digestion of prey tissue and thereby influence microbial community structure and host nutritional state.
The tendency for fish to harbor microbiota from their local prey is inconsistent with a recent experimental study of stickleback. In laboratory-reared stickleback, different foods induced divergence in microbiota composition (Bolnick et al., 2014b). However, the microbiota of fish fed Daphnia were not more similar to Daphnia microbes, nor did chironomid-fed fish carry more chironomid-associated microbes. The experimental results suggest that differential colonization of microbes is relatively unimportant compared with nutritional effects on gut microbial persistence. Our current results, using entirely natural populations and their prey, present a somewhat different but not necessarily contradictory conclusion, that microbial colonization from food does explain some of the natural among-population variation in stickleback gut microbiota.
Abiotic ecological features were also correlated with gut microbial diversity. Among-individual microbial diversity was higher in shallow and deep lakes than in lakes of intermediate depth. This association most likely arose from stickleback foraging ecology, although the direction is opposite from what we expected. This suggests that diet-based colonization alone cannot account for beta-diversity within lakes, consistent with previous field and lab studies showing that mixed-diet fish actually exhibit reduced rather than elevated microbial diversity (Bolnick et al., 2014b). In conclusion, by analyzing both host genotype and environmental microbes (from water and prey) we can confirm that stickleback gut microbial metapopulation structure is in part shaped by differential colonization from the outside environment (ingested prey and water). However, host genotype effects suggest that there is an important additional role for species sorting (differential persistence of microbe OTUs) once microbe colonists arrive in the fish gut.
Species sorting and persistence
The system of geographically close but genetically differentiated lake populations allowed us to measure the effect of genetic distance on gut microbiota composition on a small spatial scale (for example, with minimal climactic or biogeochemical heterogeneity). Genetic distance among populations correlated with gut microbiota distance. In contrast, geographic distance among the six lakes had no effect on microbial differentiation. This suggests that a substantial portion of gut microbiota variation is controlled by host genotype. Note that geographic distance did have an effect on microbial differentiation when we examine all 10 populations sampled here, but this geographic effect is confounded by environmental differences, because the two brackish sample sites are also the most distant.
Genotype-microbiota correlations became even stronger after controlling for water- and prey-associated microbes. Consequently, differential microbe exposure does not cause the strong positive association between among-population genetic and microbial divergence. We infer that colonization effects, at least as measured by water and prey microbes, are weak compared with genotype effects in microbial species sorting. Such genetic control could arise either through genetic differences in mucosal immune function (for example, MHC genotype, Bolnick et al., 2014a) or through genetically based differences in host ecology (for example, diet; Bolnick et al., 2014c).
Surprisingly, populations with greater genetic heterozygosity tended to exhibit lower among-individual microbial variation. The physiological mechanisms underlying these effects are not known at present. However, the results presented here are consistent with a recent study from a single population of stickleback, showing that individuals with more diverse Major Histocompatibility Complex class II (MHC II) alleles have less diverse gut microbiota (Bolnick et al., 2014a). We speculate that the effect of overall heterozygosity described here may be a proxy for immunogenetic diversity (possibly at the MHC), which tends to reduce microbial diversity. MHC is also known to differ among stickleback populations (Eizaguirre and Lenz, 2010; Stutz and Bolnick, 2014), and so may contribute to the genetic basis of population differences in gut microbiota. Further studies will be required to test these possibilities.
In conclusion, our work demonstrates substantial gut microbiota differentiation even among geographically nearby vertebrate populations, and that the gut microbiota is shaped by both host ecology and genetics. Using metapopulation theory as a framework, we tested whether colonization processes (input of microbes from outside sources) can explain the among-population differences in gut microbiota. We confirmed that host habitat and food-associated microbes (but not water-borne microbes) predict population differences in gut microbiota. However, host genetic divergence was a stronger predictor of microbial differences among populations, especially once food- and water-associated microbes were removed from consideration. Because host immune genotype has previously been shown to affect gut microbiota in this study organism (Bolnick et al., 2014a), we suggest that these genotypic effects are indicative of species sorting processes within the host. This inference is strengthened by our finding that genetic variation is also associated with microbial variation within populations (where water microbiota and prey microbiota are largely shared). These results represent one of the first attempts to identify and partition colonization and sorting processes in a wild gut microbiota metapopulation. Further research will be required to test additional sources of colonization, to distinguish various mechanisms of microbial sorting and to extend these approaches to additional species. Ultimately, understanding microbial metapopulation assembly rules will be necessary to guide therapeutic or agricultural manipulations of symbiotic microbiota.
References
Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H et al. (2013). Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500: 232–236.
Benson AK, Kelly SA, Legge R, Ma F, Low SJ, Kim J et al. (2010). Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci USA 107: 18933–18938.
Bergh Ø . (1995). Bacteria associated with early life stages of halibut, Hippoglossus hippoglossus L., inhibit growth of a pathogenic Vibrio sp. J Fish Dis 18: 31–40.
Biswas A, Kobayashi KS . (2013). Regulation of intestinal microbiota by the NLR protein family. Int Immunol 25: 207–214.
Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R et al. (2013). Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Method 10: 57–59.
Bolnick DI . (2011). Sympatric speciation in threespine stickleback: why not? Int J Ecol 2011: 1–15.
Bolnick D, Snowberg LK, Caporaso JG, Lauber CL, Knight R, Stutz WE . (2014a). Major Histocompatibility Complex class IIB polymorphism influences gut microbiota composition and diversity. Mol Ecol 23: 4831–4845.
Bolnick DI, Snowberg LK, Hirsch PE, Lauber PE, Knight R, Caporaso JG et al. (2014b). Individuals’ diet diversity influences gut microbial diversity in two freshwater fish (threespine stickleback and Eurasian perch). Ecol Lett 17: 979–987.
Bolnick DI, Snowberg LK, Hirsch PE, Lauber CL, Org E, Parks B et al. (2014c). Individual diet has sex-dependent effects on gut vertebrate gut microbiota. Nat Commun 5: 4500.
Caldera EJ, Bolnick DI . (2008). Effects of colonization history and landscape structure on genetic variation within and among lacustrine populations of threespine sticklebacks in a watershed. Evol Ecol Res 10: 1–24.
Campbell AC, Buswell JA . (1983). The intestinal microflora of farmed Dover sole (Solea solea) at different stages of fish development. J Appl Bacteriol 55: 215–223.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336.
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci USA 108: 4516–4522.
Carlisle EM, Poroyko V, Caplan MS, Alverdy JA, Liu D . (2011). Gram negative bacteria are associated with the early stages of necrotizing enterocolitis. PLoS One 6: e18084.
Carvaho FA, Aitken JD, Vijay-Kumar M, Gewirtz AT . (2012). Toll-like receptor-gut microbiota interactions: perturb at your own risk!. Annu Rev Physiol 74: 177–198.
Cebula A, Seweryn M, Rempala GA, Pabla SS, McIndoe RA, Denning TL et al. (2013). Thymus-derived regulatory T cells contribute to tolerance to commensal microbiota. Nature 497: 258–262.
Costello EK, Stagaman K, Dethlefsen L, Bohannan BJM, Relman DA . (2012). The application of ecological theory toward an understanding of the human microbiome. Science 336: 1255–1262.
Dethlefsen L, Eckburg PB, Bik EM, Relman DA . (2006). Assembly of the human intestinal microbiota. Trends Ecol Evol 21: 517–523.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72: 5069–5072.
Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N et al. (2010). Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci USA 107: 11971–11975.
Eizaguirre C, Lenz TL . (2010). Major histocompatibility complex polymorphism: dynamics and consequences of parasite-mediated local adaptation in fishes. J Fish Biol 77: 2023–2047.
Faith JJ, McNulty NP, Rey FE, Gordon JI . (2011). Predicting a human gut microbiota's response to diet in gnotobiotic mice. Science 333: 101–104.
Frazier TH, DiBaise JK, McClain CJ . (2011). Gut microbiota, intestinal permeability, obesity-induced inflammation, and liver injury. JPEN J Parenter Enteral Nutr 35(5 Suppl 14S–20S.
Gomez GD, Balcazar JL . (2008). A review on the interactions between gut microbiota and innate immunity of fish. FEMS Immunol Med Microbiol 52: 145–154.
Hagen DW, McPhail JD . (1970). The species problem within Gasterosteus aculeatus on the Pacific coast of North America. J Fisheries Board Can 27: 147–155.
Hansen GH, Olafsen JA . (1999). Bacterial interactions in early life stages of marine cold water fish. Microb Ecol 38: 1–26.
Haiser HJ, Turnbaugh PJ . (2012). Is it time for a metagenomic basis of theraputics? Science 336: 1253–1255.
Hendry AP, Bolnick DI, Berner D, Peichel CL . (2009). Along the speciation continuum in sticklebacks. J Fish Biol 8: 2000–2036.
Hendry AP, Kaeuffer R, Crispo E, Peichel CL, Bolnick DI . (2013). Evolutionary inferences from the analysis of exchangeability. Evolution 67: 3429–3441.
Hildebrandt MA, Hoffmann C, Sherrill–Mix SA, Keilbaugh SA, Hamady M, Chen YY et al. (2009). High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology 137: 1716–1724.
Hofer U, Speck RF . (2009). Disturbance of the gut-associated lymphoid tissue is associated with disease progression in chronic HIV infection. Semin Immunopathol 31: 257–266.
Hooper LV, Littman DR, Macpherson AJ . (2012). Interactions between the microbiota and the immune system. Science 336: 1268–1273.
Jones FC, Grabherr MG, Chan YF, Russell P, Mauceli E, Johnson J et al. (2012). The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484: 55–61.
Kovacs A, Ben-Jacob N, Tayem H, Halperin E, Iraqi FA, Gophna U . (2011). Genotype is a stronger determinant than sex of the mouse gut microbiota. Microb Ecol 61: 423–428.
Knights D, Costello EK, Knight R . (2011). Supervised classification of human microbiota. FEMS Microbiol Rev 35: 343–359.
Laparra JM, Sanz Y . (2010). Interactions of gut microbiota with functional food components and nutraceuticals. Pharmacol Res 61: 219–225.
Lavin PA, McPhail JD . (1985). The evolution of freshwater diversity in the threespine stickleback (Gasterosteus aculeatus): site-specific differentiation of trophic morphology. Can J Zool 83: 2632–2638.
Leão PN, Ramos V, Vale M, Machado JP, Vasconcelos VM . (2012). Microbial community changes elicited by exposure to cyanobacterial allelochemicals. Microb Ecol 63: 85–95.
Levins R . (1970). Extinction. Lecture Note Math 2: 75–107.
Ley RE, Lozupone CA, Hamady M, Knight R, Gordon JI . (2008). Worlds within worlds: evolution of the vertebrate gut microbiota. Nat Rev Microbiol 6: 776–788.
Liston J . (1956). Quantitative variations in the bacterial flora of flatfish. J Gen Microbiol 15: 305–314.
Lozupone C, Faust K, Raes J, Faith JJ, Frank DN, Zaneveld J et al. (2012). Identifying genomic and metabolic features that can underlie early successional and opportunistic lifestyles of human gut symbionts. Genome Res 22: 1974–1984.
Lozupone CA, Knight R . (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71: 8228–8235.
Lozupone CA, Knight R . (2007). Global patterns in bacterial diversity. Proc Natl Acad Sci USA 104: 11436–11440.
Mahowald MA, Rey FE, Seedorf H, Turnbaugh PJ, Fulton RS, Wollam A et al. (2009). Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc Natl Acad Sci USA 106: 5859–5864.
Maslowski KM, Mackay CR . (2011). Diet, gut microbiota and immune responses. Nat Immunol 12: 5–9.
Matthews B, Marchinko KB, Bolnick DI, Mazumder A . (2010). Specialization of trophic position and habitat use by sticklebacks in an adaptive radiation. Ecology 91: 1025–1034.
Meadow JF, Bateman AC, Herkert KM, O’Connor TK, Green JL . (2013). Significant changes in the skin microbiome mediated by the sport of roller derby. PeerJ 1: e53.
Muegge BD, Kuczynski J, Knights D, Clemente JC, Gonzalez A, Fontana L et al. (2011). Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 332: 970–974.
Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson GR, Wei J et al. (2012). Host-gut microbiota metabolic interactions. Science 336: 1262–1267.
O'Dwyer JP, Kembel SW, Green JL . (2012). Phylogenetic diversity theory sheds light on the structure of microbial communities. PLoS Comput Biol 8: e1002832.
Pantoja-Feliciano IG, Clemente JC, Costello EK, Perez ME, Blaser MJ, Knight R et al. (2013). Biphasic assembly of the murine intestinal microbiota during early development. ISME J 7: 1112–1115.
Parks JM, Johs A, Podar M, Bridou R, Hurt RA, Smith SD et al. (2013). The genetic basis for bacterial mercury methylation. Science 339: 1332–1335.
R Development Core Team. (2012) R: A language and environment for statistical computing. R Foundation for Statistical Computing: Vienna, Austria, ISBN 3-900051-07-0, URL http://www.R-project.org/.
Reusch TB, Wegner KM, Kalbe M . (2001). Rapid genetic divergence in postglacial populations of threespine stickleback (Gasterosteus aculeatus): the role of habitat type, drainage and geographical proximity. Mol Ecol 10: 2435–2445.
Rey FE, Faith JJ, Bain J, Muehlbauer MJ, Stevens RD, Newgard CB et al. (2010). Dissecting the in vivo metabolic potential of two human gut acetogens. J Biol Chem 285: 22082–22090.
Robertson SJ, Girardin SE . (2013). Nod-like receptors in intestinal host defense: controlling pathogens, the microbiota, or both? Curr Opin Gastroenterol 29: 15–22.
Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA et al. (2011). The toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science 332: 974–977.
Snowberg L, Hendrix KA, Bolnick DI . The magnitude of individual specialization varies across lake populations of threespine stickleback (Gasterosteus aculeatus). Oecologia; doi:10.1007/s00442-014-3200-7.
Stutz WE, Bolnick DI . (2014). Stepwise threshold clustering: a new method for genotyping MHC Loci using next-generation sequencing technology. PLoS One 9: e100587.
Sullam KE, Essinger SD, Lozupone CA, O'Connor MP, Rosen GL, Knight R et al. (2012). Environmental and ecological factors that shape the gut bacterial communities of fish: a meta-analysis. Mol Ecol 21: 3363–3378.
Turnbaugh PJ, Backhed F, Fulton L, Gordon JI . (2008). Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 3: 213–223.
Wong JM, Esfahani A, Singh N, Villa CR, Mirrahimi A, Jenkins DJ et al. (2012). Gut microbiota, diet, and heart disease. J AOAC Int 95: 24–30.
Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA et al. (2011). Linking long-term dietary patterns with gut microbial enterotypes. Science 334: 105–108.
Acknowledgements
This study was supported by funding from the Howard Hughes Medical Institute, and NSF grant DEB-1144773 to DIB.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Smith, C., Snowberg, L., Gregory Caporaso, J. et al. Dietary input of microbes and host genetic variation shape among-population differences in stickleback gut microbiota. ISME J 9, 2515–2526 (2015). https://doi.org/10.1038/ismej.2015.64
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2015.64
This article is cited by
-
Collembolans maintain a core microbiome responding to diverse soil ecosystems
Soil Ecology Letters (2024)
-
Soil physicochemical properties and microorganisms jointly regulate the variations of soil carbon and nitrogen cycles along vegetation restoration on the Loess Plateau, China
Plant and Soil (2024)
-
The characteristics of the intestinal bacterial community from Oreochromis mossambicus and its interaction with microbiota from artificial fishery habitats
BMC Ecology and Evolution (2023)
-
Co-diversification of an intestinal Mycoplasma and its salmonid host
The ISME Journal (2023)
-
The effects of host quantitative genetic architecture on the gut microbiota composition of Chinook salmon (Oncorhynchus tshawytscha)
Heredity (2023)
