
Abstract
Animals and plants are increasingly suffering from diseases caused by fungi and oomycetes. These emerging pathogens are now recognized as a global threat to biodiversity and food security. Among oomycetes, Saprolegnia species cause significant declines in fish and amphibian populations. Fish eggs have an immature adaptive immune system and depend on nonspecific innate defences to ward off pathogens. Here, meta-taxonomic analyses revealed that Atlantic salmon eggs are home to diverse fungal, oomycete and bacterial communities. Although virulent Saprolegnia isolates were found in all salmon egg samples, a low incidence of Saprolegniosis was strongly correlated with a high richness and abundance of specific commensal Actinobacteria, with the genus Frondihabitans (Microbacteriaceae) effectively inhibiting attachment of Saprolegniato salmon eggs. These results highlight that fundamental insights into microbial landscapes of fish eggs may provide new sustainable means to mitigate emerging diseases.
Similar content being viewed by others


Introduction
Fungal diseases that were previously not considered as major threats to ecosystem functioning are now causing severe ecological disruption (Fisher et al., 2012). Among the fungi, Fusarium solani is causing mass mortality in eggs of endangered sea turtles in Cape Verde (Sarmiento-Ramirez et al., 2010), and Batrachochytrium dendrobatidis and Batrachochytrium salmandrivorans are involved in major amphibian declines worldwide (Woodhams et al., 2011; Martel et al., 2013). Among the fungal-like oomycetes, Aphanomyces astaci, Aphanomyces invadans and Saprolegnia species are causing significant reductions in crayfish, fish and amphibian populations (Fernandez-Beneitez et al., 2008; Phillips et al., 2008; Krugner-Higby et al., 2010; Bruno et al., 2011; Van den Berg et al., 2013), respectively. Saprolegniosis is a major disease problem in different wild and farmed fish species, including salmonid species such as Atlantic salmon, rainbow and brown trout, and non-salmonid species including perch, eels and catfish (Bruno et al., 2011). This disease is characterized by white and grey patches of mycelial growth on the skin and fins of adult fish, and cotton-like filamentous mycelium on eggs. In fish, death often occurs from disruption of the osmotic balance (haemodilution) following destruction of large areas of the epidermis by massive, invasive hyphal growth (Bruno et al., 2011). Fish eggs, on the other hand, are thought to be killed by hyphal breaching of the chorionic membrane regulating the osmosis of the embryo. Saprolegnia species can produce flagellated zoospores to disperse in the aquatic environment. Saprolegnia also forms secondary zoospores and secondary cysts that, for some species, contain boathooks that are presumed to aid in attachment to the fish skin or promote floating (buoyancy) in water (Van den Berg et al., 2013). Losses resulting from Saprolegniosis average 10% in eggs and young fish, but losses of up to 50% have been reported (Hatai and Hoshiai, 1992, 1994; Bruno et al., 2011; Van den Berg et al., 2013).
To control and prevent the spread of emerging pathogens, several conservation and disease mitigation strategies have been proposed because a singular solution is doubtful given the rich diversity of fish and amphibian habitats (Woodhams et al., 2011). In aquaculture, immunization and chemical control are among the preferred approaches to mitigate diseases (Bruno et al., 2011; Van den Berg et al., 2013). For Saprolegniosis, however, no effective immunization is currently available and nonspecific chemical agents such as malachite green and formalin have been or will be banned, leading to a dramatic re-emergence of Saprolegniosis. Habitat bioaugmentation and introduction of protective microbiota have been proposed as potential strategies to rescue and protect fish and amphibians from emerging disease. Over the past years, several studies have unequivocally shown that microbes play a pivotal role in the protection of eukaryotes (humans, plants and insects) against pathogens (Round et al., 2010; Mendes et al., 2011; Weiss and Aksoy, 2011; Everard et al., 2013; Stecher et al., 2013; Turner et al., 2013). For fish and amphibians, microbiome studies have only been initiated recently (Cahill, 1990; Hansen and Olafsen, 1999; Schulze et al., 2006; McKenzie et al., 2012). Several studies have indicated that specific bacteria can contribute to the protection of fish and amphibians against fungal and oomycete pathogens. This was shown for Janthinobacterium lividum, a bacterium that was isolated from frog skin and provided protection against B. dendrobatidis (Harris et al., 2009).
Here, we deciphered the microbiome of Atlantic salmon eggs and determined how Saprolegniosis affects the structure (richness, evenness) of fungal, oomycete and bacterial communities, with the ultimate goal to identify commensal microbes that prevent or delay disease onset and/or development.
Materials and methods
Salmon egg sample collection for microbial community analysis
Atlantic salmon (Salmo salar L.) eyed eggs and the incubation water from each of the tanks were collected separately from a commercial hatchery in Scotland. The eggs were spawned in November 2010 and incubated in a flow-through system at a flow rate of 25–30 l s−1 at 3–5 °C. The water source for the incubation of the salmon eggs is a catchment area that is an upland peat moor with conifer plantations, grazing cattle, sheep and wild animals, ∼3 km from the hatchery and is delivered via a hydroelectric scheme using a hill loch as a source. Before the water enters the egg incubators, it is filtered through a 10-μm filter and a carbon filter and is subsequently treated with ultraviolet light for a few seconds. Eggs received a 1-h 1000 p.p.m. formalin bath treatment weekly, and were collected on 14 February 2011. For more details on each of the 12 egg samples, see Supplementary Table 1. The microbial community was obtained by shaking the eggs and incubator water at 180 r.p.m. at 6 °C for 12 h, and subsequent centrifugation of the water at 24 000 × g at 4 °C for 20 min. Cell pellets were resuspended in sterile demineralized water, concentrating the original sample 30–80 times, and stored at −80 °C before DNA isolation. One aliquot was stored at −80 °C in 20% (v/v) glycerol for bacterial isolation (Supplementary Figure 1).
Scanning electron microscopy
A selection of salmon egg samples showing visual signs of Saprolegnia infection were collected and stored in a 1% gluteraldehyde and 4% formalin solution. Subsequently, samples were fixed in 2.5% gluteraldehyde and then treated with 1% osmium tetroxide for 1 h. Samples were dehydrated with alcohol and subsequently dried using hexamethyldisilazane. Dried samples were coated in gold using an EMitech Limited (Ashford, UK) K550 sputter coater. Coated samples were viewed in a Zeiss (Oberkochen, Germany) EVO MA10 scanning electron microscope at 10 kV.
Metagenomic DNA isolation and storage
DNA was isolated from the pellet suspensions (described above) in duplicate with PowerSoil DNA isolation kit (MO BIO Laboratories, Inc., Carlsbad, CA, USA) according to the manufacturer’s instructions. Duplicate DNA extracts were pooled and an aliquot was analysed on a 0.8% (w/v) agarose gel with SmartLadder MW-1700-10 (Eurogentec, Seraing, Belgium) as a size marker. The DNA concentration was measured with a NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA, USA). DNA samples were stored at −80 °C.
PhyloChip analysis
PhyloChip analysis of the bacterial and archaeal community structure of each sample (∼0.5 μg DNA) was conducted at Second Genome (South San Francisco, CA, USA). Sample and data processing and data reduction were performed as described previously (Hazen et al., 2010). Annotation of operational taxonomic units (OTUs) was performed based on the Greengenes database (McDonald et al., 2012). Cluster analysis with Bray–Curtis similarity measures was performed in Primer-E v6.1.13 (Primer-E Ltd, Ivybridge, UK). Nonmetric multidimensional scaling and analysis of similarity (Ramette, 2007) were performed in Paleontological Statistics freeware package (PAST v2.10, http://folk.uio.no/ohammer/past/) (Hammer et al., 2001).
Clone library sequencing of oomycete and fungal communities
The internal transcribed spacer (ITS) region of fungal and oomycete rRNA genes was PCR amplified with primers ITS1F and ITS4 (White et al., 1990; Gardes and Bruns, 1993), and Oom-Up5.8S (5′-TGCGATACGTAATGCGAATT-3′) and Oom-Lo28S (5′-ACTTGTTCGCTATCGGTCTCGCA-3′) (Mazzola et al., 2002; Tambong et al., 2006), respectively. Amplification reactions were conducted in 50 μl volumes with Taq DNA polymerase (Invitrogen, Grand Island, NY, USA) according to the manufacturer’s instructions with 1 μl DNA (5–10 ng), 1 cycle at 94 °C for 3 min, 40 cycles at 94 °C for 1 min, 50 °C for 45 s, 72 °C for 1 min and 1 cycle at 72 °C for 7 min. The dominant representatives of fungal and oomycete populations were determined by sequencing clone libraries. Amplicons were cloned into the TOPO-TA vector and transformed into chemically competent Escherichia coli Top10 cells according to the manufacturer’s protocol (Invitrogen). Inserts were PCR amplified from intact cells using primer pair M13F/M13R and purified with ExoSAP-IT (USB, Cleveland, OH, USA) according to the manufacturer’s instructions. A total of 20 and 5–6 clones per sample were selected for fungal and oomycete amplicon sequencing, respectively, with primer T7 and GenomeLab DTCS Quick Start Kit using the Beckman Coulter CEQ 8000 Genetic Analysis system (Beckman Coulter, Brea, CA, USA). Sequences of at least 400 bp were analysed by BLASTn (Altschul et al., 1990) on the National Center for Biotechnology Information (NCBI) database website (www.ncbi.nlm.nih.gov/). A cutoff of >93% identity and a 400-bp alignment length were used for identification (Pounder et al., 2007; Nilsson et al., 2008).
T-RFLP analysis of fungal community
Fluorescently labelled PCR products of the ITS region were generated using the D4-labelled ITS1F primer in conjunction with D3-labelled ITS4 primer. PCR was performed in quadruplicate 25 μl reactions containing 1 × Promega PCR Master mix (Promega, Madison, WI, USA), 200 mM primer, 4 μg μl−1 bovine serum albumin and 0.1–0.5 μl DNA template (0.5–5 ng) with above-mentioned cycling conditions. PCR products were digested in duplicate reactions with 10 U of the restriction endonucleases HaeIII and HhaI at 37 °C for 6 h. Restriction enzymes were heat inactivated at 80 °C for 10 min. Restriction fragments were purified by ethanol precipitation and fragments were resuspended in 40 μl of sample loading solution and 0.25 μl 600 size standards. Terminal restriction fragment length polymorphism (T-RFLP) fragments were separated using the CEQ 8000 Genetic Analysis System with denaturation for 120 s at 90 °C, injection for 15 s at 2 kV, separation for 90 min at 4.8 kV and a capillary temperature of 50 °C. T-RFLP profiles were analysed using the Fragment Analysis Module of the CEQ 8000 Genetic Analysis Software, with a slope threshold of 10, 1% relative peak height threshold, 95% confidence level, quartic calibration curve with PA, ver. 1, dye mobility calibration and calculated dye spectra. The baseline value was set to exclude background peaks that commonly occurred in tandem with the DNA standard peaks. Fragments were binned with a 1-bp margin using the AFLP tool in the Fragment Analysis software. Profiles that consisted of mean fragment length and fluorescence intensities were generated for each sample. As different amounts of DNA were used for amplification between samples, no accurate analysis of T-RF abundance could be carried out and we analysed the data only on the presence/absence of T-RF peaks. Cluster analysis was performed as mentioned above.
Isolation of oomycetes and fungi from salmon eggs
Per sample, one to two salmon eggs were placed on potato dextrose agar (PDA, Oxoid, Hampshire, UK). The cultures were incubated for 2 weeks at room temperature (20±1 °C) and for 17–18 days at 18 °C. The oomycete and fungal isolates were purified by subculturing hyphal tips three times on fresh PDA supplemented with 100 μg ml−1 streptomycin and 50 μg ml−1 tetracycline.
ITS sequencing and phylogenetic analysis of oomycete isolates
For genomic DNA extraction, hyphae were harvested in 90 μl of 0.5 M NaOH and disrupted by Mixer Mill MM400 (Retsch, Haan, Germany) at a frequency of 30/s for 35 s in the presence of one sterile glass bead. The tube was placed at room temperature for 30 min to 24 h until complete cell wall disruption. The hyphal suspension was diluted with sterile 0.1 M Tris-HCl (pH 7.0) and ITS rRNA genes were amplified with ITS1 and ITS4 primers in 25 μl volumes, each consisting of 2 μl of diluted DNA template with GoTaq DNA polymerase (Promega) according to the manufacturer’s protocol. The amplification program consisted of 1 cycle at 94–95 °C for 2–5 min, 30–35 cycles at 94 °C for 1 min, 55 °C, 60 °C or 65 °C for 30–60 s, 72 °C for 30–120 s and 1 cycle at 72 °C for 5–10 min. PCR products were sequenced by Macrogen Inc. (Amsterdam, The Netherlands). Contigs were created from the sequence fragments using Geneious v6.1.4 (Kearse et al., 2012) and aligned using MAFFT v7.0 with G-INS-i algorithm using default settings (Katoh and Standley, 2013), including previously identified reference sequences (Sandoval-Sierra et al., 2013). Two different approaches were applied: Bayesian inferences and Maximum Likelihood inference. Bayesian inference analysis with Markov-chain Monte Carlo, MrBayes v3.2.1 (Ronquist et al., 2012), was run at temperature parameter of 0.2, sample frequency of 1000 and three independent runs with two hot and six cold chains each for 20 million generations. Estimation of the effective samples size in the Markov-chain Monte Carlo and determination of a suitable burn-in time was checked with Tracer v1.5 (Rambaut and Drummond, 2007). A burn-in of 2 million generations was discarded after checking for stability on the log-likelihood curves, and the remaining trees from the independent runs were combined to build a 50% majority-rule consensus tree. The evolutionary model used for the molecular data in MrBayes was HKY+G and was obtained by running the data sets in jModelTest 2 (Darriba et al., 2012). Maximum Likelihood inference analysis was carried out with RaxmlGUI v7.4.2 (Silvestro and Michalak, 2012) with a random starting tree, included the GTRGAMMA option and employed the rapid hill-climbing algorithm (Stamatakis et al., 2007). Clade support was assessed with 1000 bootstrap replicates, with the rapid-hill climbing algorithm (Stamatakis et al., 2008). Phylogram trees were visualized with FigTree (tree.bio.ed.ac.uk/software/figtree/).
Isolation, in vitro activity and identification of Actinobacterial isolates
Cell pellets obtained from the salmon eggs samples and stored in 20% glycerol at −80 °C were resuspended in sterile demineralized water and dilution plated on humic acid-vitamin (HA) and starch agar (GA) modified from Zhang (1990). Plates were incubated at 30 °C and bacterial colony-forming units were determined after 6 weeks. HA medium, modified from Hayakawa and Nonomura (1987), contained 1.0 g l−1 humic acid (dissolved in 10 ml of 0.2 N NaCl), 0.5 g l−1 Na2HPO4, 1.71 g l−1 KCl, 0.05 g l−1 MgSO4.7H2O, 0.01 g l−1 FeSO4.7H2O, 0.02 g l−1 CaCO3 and 18.0 g l−1 agar. Starch agar (GA) (Zhang, 1990) contained 15 g l−1 potato starch (soluble starch), 0.5 g l−1 KNO3, 0.5 g l−1 K2HPO4, 0.5 g l−1 MgSO4.7H2O, 5.0 g l−1 NaCl, 5.0 g l−1 KCl, 0.01 g l−1 FeSO4.7H2O and 18.0 g l−1 agar. Both HA and GA were pH adjusted to 7.0–7.4 and were supplemented with 1 ml l−1 of B vitamin solution, 20 μg ml−1 nalidixic acid, 20 μg ml−1 Trimethoprim and 100 μg ml−1 Delvocid after autoclaving. B vitamin solution (Nonomura and Ohara, 1969) (suspended in Milli-Q water) included 0.5 mg ml−1 of thiamine hydrochloride, riboflavin, nicotinic acid, pyridoxine hydrochloride, myo-inositol, calcium pantothenate and 4-aminobenzoic acid and 0.25 mg ml−1 D(+)-biotin. From each sample, 40 single colonies were randomly isolated at different time points from both media and purified on GA or tryptone soya broth (Oxoid) supplemented with 10% sucrose (TSBS) with or without 1.8% (w/v) agar. Isolates were stored at −20 °C and −80 °C in 20–40% glycerol. In vitro antagonistic activity of the isolates against Saprolegnia diclina 1152F4 was determined by placing an agar plug of each Actinobacterial isolate at the edge of 1/5th strength PDA plates. After incubation at 30 °C for 7–10 days, an agar plug from a fresh S. diclina 1152F4 culture was placed in the centre of the plate. Plates were incubated for 4–5 days at 25 °C and inhibition of hyphal growth was scored. For DNA isolation, Actinobacterial cells were disrupted by incubation of 50 μl TSBS culture at 95 °C for 10 min and PCR was performed with Actinobacterial-specific primers Com2xf (5′-AAACTCAAAGGAATTGACGG-3′) and Ac1186r (5′-CTTCCTCCGAGTTGACCC-3′) (Schäfer et al., 2010). Amplification reactions were conducted in 25 μl volumes using GoTaq DNA polymerase (Promega) according to the manufacturer’s instructions, with 1 cycle at 95 °C for 3 min, 35 cycles of 94 °C for 30 s, 60 °C for 30 s, 72 °C for 30 s and 1 cycle at 72 °C for 15 min. All isolates positive for the amplified fragments were sent for 16S rRNA sequencing by Macrogen Inc. Sequence identity was obtained by comparison with sequences deposited in the Greengenes database (McDonald et al., 2012) (http://greengenes.lbl.gov/) and Ribosomal Database Project (Cole et al., 2009) (RDP, http://rdp.cme.msu.edu/). Among the 526 Actinobacterial OTUs with significant differential abundance based on the PhyloChip analyses, the 16S rRNA sequences (⩾850 bp) of 413 OTUs were downloaded from the Greengenes database. These sequences were subjected to phylogenetic analysis with ClustalW in MEGA5 (Tamura et al., 2011) to generate a neighbour-joining (Saitou and Nei, 1987) consensus tree with 1000 bootstrap replicates (Felsenstein, 1985). The evolutionary distances were analysed by Tamura–Nei model (Tamura and Nei, 1993) and the evolutionary rates among sites were modelled by a gamma distribution.
Salmon eggs for in vivo experiments
Atlantic salmon (Salmo salar L.) eyed eggs of strain AquaGen Atlantic QTL-innOva IPN from AquaGen AS (Trondheim, Norway) were used for the in vivo experiments. Salmon egg ages at the day of shipment were as follows: for the pathogenicity assay of Saprolegnia isolates, the eggs were 385 degree-days; for the in vivo assays with the Actinobacterial isolates, the eggs were 320 degree-days. All egg batches were disinfected during incubation and before transport with buffodine (1:100, 10 min) and treated with formalin according to Aquagen’s in-house protocols. Upon arrival, blank, pin-eyed and white eggs were removed if observed.
Preparation of Saprolegnia inocula and pathogenicity assays
Live eggs were placed in 97–98 °C sterile distilled water or well water and incubated for 80–150 s. The resulting dead eggs were drained and placed on PDA agar plates overgrown with Saprolegnia isolates and incubated overnight at 25 °C. Each egg was then transferred into a well of a 24-well cell-culture plate (Greiner Bio-One, Kremsmünster, Austria) and further incubated for 1 day at 15–25 °C until Saprolegnia hyphal colonization was visible. The slow-growing Saprolegnia isolates 746F3 and 736F2 were further incubated for 2 and 4 days, respectively. After overnight acclimatization of the live eggs, two Saprolegnia-infected dead salmon eggs (inocula) were added to each cup containing 30 live eggs in a similar setup as described in Figure 4c, with the exception that here 3 l of dechlorinated Norwegian tap water, and not well water, was used. Isolate 736F2 was added to the salmon eggs 2 days later as this isolate was a slow grower. Each treatment consisted of an incubator containing three cups with 30 salmon eggs each. In the control treatment no Saprolegnia was added. The water temperature was maintained at 10±1 °C by adding ice blocks into each incubation unit every 12 h. After 8 days post inoculation(6 days post inoculation for isolate 736F2), the mortality percentage of the salmon eggs was determined visually.
In vivo bioassays to test disease suppression by Actinobacteria
The incubation setup (Supplementary Figure 4) was located in a ventilated room of constant 15 °C ambient temperature. The egg incubation units, containing 2 l of well water (Supplementary Table 6), were randomly distributed in an incubation tank with circulating water of 5–7 °C. Polystyrene panels were used to cover the incubation tank to maintain darkness and insulation without blocking aeration. Each incubation unit was aerated with an air stone connected to an air pump. Dissolved oxygen levels and temperature, measured with a pH/Oxi 340i multi-parameter instrument (WTW GmbH, Weilheim, Germany), ranged from 101.8% to 103.4% and from 5.7 °C to 6.4 °C, respectively. Within each egg incubation unit, 51±1 live eggs were placed in perforated plastic cups in triplicate. The cups were stabilized by placing a Petri dish (145 mm diameter) and a weight on top. The eggs were introduced into each incubation unit immediately after arrival from Norway and allowed to acclimatize and rehydrate overnight.
Actinobacterial isolates were inoculated in TSBS and incubated at 30 °C at 200 r.p.m. for 2–4 days. Cultures were centrifuged at 3400 × g for 5–15 min at room temperature. The cell pellets were washed and resuspended in sterile well water and OD600 values determined by spectrophotometry for the Frondihabitans sp. and Arthrobacter sp. For the Streptomyces isolates, the OD600 could not be determined because of aggregation of cells. Approximate equal amounts of cells were determined based on an estimate of the size of the cell pellet after centrifugation. On day 1, the Frondihabitans sp. and Arthrobacter sp. were added to the incubation units at a final concentration of 107 cells per ml or 0.5 ml and 1.5 ml of Streptomyces sp. 1 and 2, respectively. In the consortium of all four isolates, one quarter of the cell suspensions of each bacterium was added. After 3 days of incubation, two S. diclina 1152F4 inocula were placed at opposite sides in each cup. As control treatments, salmon eggs were treated with S. diclina only or with malachite green and S. diclina. Each treatment was conducted in duplicate, that is, two independent experimental incubation units with three technical replicates per incubation unit. For those eggs that received malachite green treatment, the eggs were placed in 1.5 l of 2 p.p.m. malachite green solution for 60–70 min with aeration. Subsequently, eggs were gently rinsed in 500 ml fresh well water and placed back into their incubation units. This was repeated every 4 days, replacing the incubation unit water with fresh well water. Hyphal expansion was photographed and assessed every other day. Based on the photos of each group of eggs, ImageJ v1.44n (Schneider et al., 2012) was used to measure the diameter of hyphal expansion. As a proxy for infection of salmon eggs by Saprolegnia, we determined hyphal attachment by lifting infection inocula with a pair of tweezers and counting the number of eggs attached to the hyphal patch (Supplementary Movie 1). Hyphal attachment was determined at day 14 (10 days post inoculation) when attachment in the control, S. diclina only, was >50% (Supplementary Table 5).
Nucleotide sequence accession numbers
The sequences of the 16S rRNA sequences of Frondihabitans sp., Arthrobacter sp. and Streptomyces sp. 1 and 2 have been deposited in GenBank under accession numbers KF741274, KF741275, KF741276 and KF741277, respectively.
Colonization of Actinobacteria on the egg surface
Colonization of the applied Actinobacterial isolates on the salmon egg surfaces was determined by harvesting one to two eggs from each cup at 0 and 10 days post inoculation, rinsing with 1 ml of sterile well water three times, draining briefly on sterile filter paper and rolling gently over the surface of HA and GA agar plates (Supplementary Movie 2). All plates were incubated at 30 °C for 5–6 weeks, and 2–8 colonies that were morphologically similar to each of the four isolates used for inoculation were purified and subjected to 16S rRNA sequencing.
Statistical analyses
For each experiment, obtained data were tested for normal distribution and homogeneity of variances before conducting parametric or nonparametric statistical analyses. Disease incidence of salmon egg samples was analysed with Mann–Whitney U-test (P<0.05). The pathogenicity of Saprolegnia isolates were compared with one-way analysis of variance and with post hoc Dunnett analysis (P<0.05). Cluster analysis and nonmetric multidimensional scaling and analysis of similarity of T-RFLP-derived data for the fungal community and PhyloChip-derived data for the bacterial and archaeal communities were conducted using Bray–Curtis similarity measures in Primer-E and in PAST. The differences in total number of bacterial and archaeal OTUs associated with diseased versus healthy eggs was analysed by Student’s t-test (P>0.05). Phylogenetic groups containing OTUs that have a significant differential abundance between diseased and healthy eggs were identified with Student’s t-test (P<0.005) in Microsoft Excel. Percentage of hyphal attachment was analysed with Kruskal–Wallis one-way analysis of variance (P<0.05). Area under hyphal expansion curve and area under hatching percentage curve of different treatments in the bioassay were compared with Kruskal–Wallis one-way analysis of variance (P<0.05). All statistical analyses were performed in IBM SPSS Statistics v20 (Armonk, NY, USA), unless mentioned otherwise.
Results and discussion
We conducted a meta-taxonomic microbiome analysis of 12 salmon egg batches from a Scottish salmon hatchery (Supplementary Figure 1 and Supplementary Methods). In hatcheries, certain salmon egg batches become infected with Saprolegnia, whereas others remain unaffected. Here, six of the sampled egg batches were selected based on a high incidence of Saprolegniosis and six based on a low disease incidence (Figure 1 and Supplementary Figure 1). From this point onwards, these two distinct sample sets are referred to as ‘diseased’ and ‘healthy’, respectively. Both sets, containing eggs from the same broodstock, received water from the same source, were all incubated at average temperatures ranging from 3 °C to 5 °C, and were all sampled at ∼85 days after fertilization (Supplementary Table 1). Total DNA was extracted from the microbiota present in the pellets obtained from each of the 12 egg samples and subjected to meta-taxonomic analyses (Supplementary Figure 1 and Supplementary Methods).

Saprolegniosis in Atlantic salmon (Salmo salar L.) eggs. (a) Diseased and healthy salmon eggs collected from a hatchery (N=6). (b) Close-up picture of diseased and healthy salmon egg. (c) Scanning electron microscopy (SEM) image of Saprolegnia-infected salmon egg showing Saprolegnia hyphae (white arrows) and bacteria (orange arrows) covering the salmon egg surface. (d) Disease incidence of salmon egg samples collected in the hatchery. Error bars indicate s.e.m. (N=6). *Statistically significant difference compared with the diseased samples. The full colour version of this figure is available at ISMEJ online.
Clone library sequencing of oomycete-specific ribosomal ITS amplicons showed the presence of Saprolegnia, Achlya and Aphanomyces (all belonging to the order Saprolegniales) in both diseased and healthy samples (Figure 2a). Achlya species have been described for diseased fish in natural environments (Jeney and Jeney, 1995; Kales et al., 2007; Sosa et al., 2007), and A. invadans is a notorious pathogen of different natural and farmed fish species worldwide, except in Europe and South America (Oidtmann, 2012). Both Achlya and Aphanomyces spp. have been isolated from eggs of sea trout (Salmo trutta m. trutta) in Polish rivers (Czeczuga et al., 2005) and from eggs of salmonids in Japan (Kitancharoen and Hatai, 1997). To date, however, no reports have indicated that Achlya or Aphanomyces are pathogenic for Atlantic salmon. Achlya or Aphanomyces were not present among the population of oomycetes isolated from the salmon eggs. Therefore, their effects on salmon eggs could not be resolved.

Oomycete community on diseased and healthy salmon eggs. (a) Number of clone library sequences obtained with oomycete-specific ITS primers. (b) Phylogenetic tree of ITS sequences of isolated oomycete strains from diseased (●) and healthy (○) salmon eggs (left). Bayesian posterior probabilities are shown at nodes followed by estimates of bootstrap support. Pathogenicity (expressed as % egg mortality (right)) of Saprolegnia isolates obtained from diseased and healthy salmon eggs. Strains that do not have a bar were not tested. Error bars indicate s.e.m. (N=3). *Statistically significant difference compared with the untreated control. R, reference strains.
For Saprolegnia species, a similar number of sequenced clones was found in diseased and healthy eggs (52% and 50%, respectively; Figure 2a). To better characterize the Saprolegnia species, eggs from both sample sets were placed on agar media to allow outgrowth of the oomycetes. Subsequent purification and ITS sequencing of 31 randomly selected oomycete isolates showed that the majority were S. diclina, with no distinct phylogenetic differences between the Saprolegnia isolates from diseased and healthy samples (Figure 2b). Furthermore, when Saprolegnia isolates from the different phylogenetic clades were tested for pathogenicity on salmon eggs, equal levels of mortality were observed for eggs inoculated with Saprolegnia isolates from diseased or from healthy egg samples (Figure 2b). Collectively, these results indicate that the difference in incidence of Saprolegniosis between the two sets of salmon egg samples from the commercial hatchery cannot be explained by differences in population structure or pathogenicity of the Saprolegnia species present in both sample sets.
Hierarchical cluster analyses of the T-RFLP data revealed no distinct differences between the fungal community from diseased or healthy salmon eggs (Figure 3b and Supplementary Figure 2a). Further ITS-clone library sequencing showed that the fungal community associated with diseased and healthy salmon egg samples was dominated by the Ascomycota (Figure 3a), mainly Microdochium (Supplementary Table 2). Microdochium species, known as snow moulds (Matsumoto, 2009), are pathogenic to cereals but also contain endophytic members that may have beneficial effects on plants (Ernst et al., 2011). For aquatic ecosystems, however, no records exist of the presence of Microdochium. Chytriomyces was only detected in the diseased egg samples, and Mortierella and Microdochium species were more abundant in diseased samples (Supplementary Table 2). These fungi have not been previously associated with salmon eggs or with Saprolegniosis. In addition, Spirosphaera and Saccharicola, the fungal genera that were only detected in healthy egg samples, have not yet been described for salmon eggs. Spirosphaera spp. are known to decompose submerged plant litter (Voglmayr, 2004), whereas some Saccharicola species (S. bicolor) cause disease in sugarcane (Eriksson and Hawksworth, 2003).

Fungal, bacterial and archaeal community analyses by clone library sequencing, T-RFLP and PhyloChip analyses. (a) Relative abundance of fungal phyla from Saprolegnia diseased and healthy salmon eggs as determined by sequence analysis of clones containing inserts of the ITS region. (b) Cluster analysis (Bray–Curtis similarity) of T-RFLP-derived data for the fungal community associated with diseased (●) and healthy (○) salmon eggs. (c) Number of OTUs of bacterial and archaeal taxa in diseased and healthy salmon egg samples as determined by number of OTUs passing stage 1 and 2 PhyCA. Error bars indicate s.e.m. (N=6). (d) Cluster analysis (Bray–Curtis similarity) of bacterial and archaeal OTUs associated with diseased (●) and healthy (○) salmon egg samples (N=6). (e) Phylogenetic groups containing OTUs that have a significant differential abundance between diseased and healthy salmon eggs.
PhyloChip (DeAngelis et al., 2009; Hazen et al., 2010)-based meta-taxonomic analysis of the bacterial community revealed a total of 31 278 bacterial and 3 archaeal OTUs. Although 16S rDNA amplicon pyrosequencing is less biased towards detecting unknown species (DeSantis et al., 2007; Kunin et al., 2010), the PhyloChip technique allows detection of a large number of bacterial taxa including less abundant taxa. A study on lignin-degrading microbes showed that the microbial community profiles obtained by the PhyloChip technique and pyrosequencing were well comparable (DeAngelis et al., 2011). The overall distribution of the four most dominant bacterial phyla for the salmon egg samples ranged from 9% for the Bacteroidetes to 12%, 24% and 44% for the Actinobacteria, Firmicutes and Proteobacteria, respectively (Figure 3c). A number of OTUs (4684) were exclusively found in healthy samples, especially taxa belonging to the phylum Actinobacteria, that is, Streptomycetaceae, Microbacteriaceae and Micrococcaceae (Figure 3c and Supplementary Table 3). Along with differences in richness, the relative abundance of the detected bacterial taxa also resulted in a clear discrimination between the diseased and healthy samples, except for sample 12 (healthy) that appeared to be more distinct from all other (healthy) samples (Figure 3d and Supplementary Figure 2b). A total of 4029 OTUs were significantly different in abundance between the diseased and healthy egg samples (Figure 3e). OTUs that were significantly more abundant in the diseased samples belonged to the Enterobacteriales, Clostridiales, Alteromonadales, Vibrionales and Aeromonadales (Figure 3e) and comprised several bacterial pathogens, including Vibrio spp. (Frans et al., 2011), Aeromonas spp. (Beaz-Hidalgo and Figueras, 2013) and Yersinia spp. (Tobback et al., 2007) that are common to salmon and other fish species. It is generally believed that Saprolegnia behaves as an opportunistic pathogen, infecting upon injury, stress or other infections (Stueland et al., 2005; Bruno et al., 2011). However, several papers have also reported that Saprolegnia acts as a primary infection agent in eggs and adult fish (Bruno et al., 2011). Thus, whether these potentially pathogenic bacterial taxa were initially present and predisposed the eggs for Saprolegnia infection or infection of the salmon eggs by Saprolegnia led to proliferation of these opportunistic pathogenic bacteria remains to be resolved.
Most of the OTUs that were significantly more abundant in healthy samples belonged to the orders Actinomycetales and Burkholderiales (Figure 3e). In fungal gardens of ants, Actinobacteria were identified that protect the ant colony from a parasitic black fungus (Phialophora sp.) (Caldera et al., 2009). Actinobacteria are prolific producers of an array of antimicrobial compounds and have been suggested as probiotic agents in aquaculture (Verschuere et al., 2000; Das et al., 2008; Dharmaraj, 2010). To study the potential role of the differentially abundant Actinobacterial taxa in protection of salmon eggs against Saprolegnia infections (Figure 3e), a total of 354 Actinobacteria were isolated from the diseased and healthy egg samples on two different media that are semiselective for Actinobacteria (Supplementary Table 4). Isolates were subcultured, purified, characterized by 16S rRNA sequencing and tested for in vitro and in vivo activity against S. diclina. Most of the cultured isolates belonged to the families Microbacteriaceae, Micrococcaceae and Streptomycetaceae and, considering their 16S rRNA-based phylogeny, were in the same clades as the differentially abundant OTUs detected by the culture-independent PhyloChip analysis (Supplementary Figure 3). OTUs detected only by PhyloChip and not by isolation belonged to Corynebacteriaceae, Dermabacteraceae, Thermomonosporaceae and Streptosporangiaceae among others.
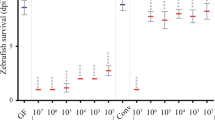
The results of the in vitro dual culture plate assays showed that 6.9% and 12.3% of the Actinobacterial isolates from diseased and healthy samples, respectively, inhibited hyphal growth of S. diclina. For the in vivo assays with salmon eggs, we selected four bacterial isolates from the obtained Actinobacterial collection belonging to clades with OTUs that were significantly more abundant in the healthy salmon egg samples (Figure 3e and Supplementary Figure 3). Two isolates belonged to the genera Frondihabitans (Microbacteriaceae) and Arthrobacter (Micrococcaceae), and two isolates to Streptomyces (Streptomycetaceae) (Figure 4a). The two Streptomyces isolates inhibited in vitro hyphal growth of S. diclina, whereas Arthrobacter and Frondihabitans were not inhibitory (Figure 4b). For the in vivo assays, salmon eggs were transferred to aerated cups inside a unit cooled to 5–7 °C and preincubated for 3 days with each of the bacterial isolates at an initial density of ∼107 cells per ml. Two heat-killed eggs overgrown with S. diclina were then placed in each cup serving as the inoculum source for pathogen outgrowth and attachment (Figure 4c and Supplementary Figure 4). After 10 days of incubation, none of the four selected Actinobacterial isolates reduced radial outgrowth of S. diclina from the inoculum source (Supplementary Figure 5a). However, the Frondihabitans isolate significantly inhibited hyphal attachment to the salmon eggs (Figure 4d and Supplementary Movie 1), even under high disease levels where malachite green treatment did not provide 100% control (Figure 4d). This suppressive effect was not observed for the Streptomyces and Arthrobacter isolates. When a consortium of all four isolates was tested, hyphal attachment was inhibited to the same level as obtained with the Frondihabitans isolate alone (Figure 4d). Subsequent analysis of the bacterial community associated with the salmon eggs (see Supplementary Movie 2) showed that, based on colony morphology and 16S rRNA sequencing, the majority of the obtained colonies were indeed the Actinobacterial isolates initially applied to the salmon eggs. In the absence of S. diclina, stress-associated hatching (Czerkies et al., 2001) of the salmon eggs was observed for one of the Streptomyces isolates but not for Frondihabitans, Arthrobacter or other Streptomyces isolates (Supplementary Figures 5b and c). None of the bacterial isolates exhibited adverse effects on alevin survival or morphology (data not shown). Collectively, these results highlight that insights into the microbial landscapes of salmon eggs allows for a taxon-targeted selection of specific commensal bacteria from fish eggs with the potential to mitigate Saprolegniosis.

Characteristics and in vivo testing of selected Actinobacterial isolates to control Saprolegniosis on salmon eggs. (a) Morphology of Frondihabitans, Arthrobacter and two Streptomyces isolates. (b) In vitro antagonism against Saprolegnia diclina 1152F4. (c) Setup of in vivo bioassay. Eggs were inoculated with bacteria and after 3 days of incubation, two S. diclina-infected salmon eggs were added at opposite ends of each cup. The diameter of hyphal expansion was determined every 2 days as indicated by the double arrow. (d) Percentage of eggs to which Saprolegnia hyphae were attached at 10 days post inoculation of the pathogen (see Supplementary Movie 1). Error bars indicate the s.e.m. (N=6). *Statistically significant difference with the control, S. diclina only (Kruskal–Wallis, P<0.05).
Frondihabitans (previously named Frondicola) was originally isolated from fallen leaf litter from a pine forest in Australia (Zhang et al., 2007; Greene et al., 2009) and has also been isolated from lichens (Cardinale et al., 2011) and the rhizosphere of Peucedanum japonicum (Lee, 2010). However, association with freshwater environments has not previously been reported. The means by which Frondihabitans limits hyphal attachment of S. diclina to the salmon eggs is not known. The observation that hyphal expansion of the pathogen from the inoculum sources was not inhibited by the Frondihabitans isolate (Supplementary Figure 5a) and that the bacterium did not show in vitro antagonism (Figure 4b) suggests that antibiosis via (water-soluble) secondary metabolites is most likely not a key mechanism of pathogen control. Microscopic analysis further demonstrated that zoospores of S. diclina were not negatively affected by the presence of Frondihabitans sp. However, the role of zoospores in pathogenesis is unclear as no zoospore-initiated secondary infection was observed throughout the experiment. Successful colonization of salmon egg surfaces by the Frondihabitans isolate may point to competitive niche exclusion as one of the underlying mechanisms of disease suppression.
The impact of the microbiota on host health and development is gaining increasing attention. For humans, environmental changes or infections can substantially influence the gut microbiome by causing blooms of microbes that are otherwise present at low abundance (Stecher et al., 2013). Human gut microbiomes can significantly drive or suppress disease development (Round et al., 2010; Everard et al., 2013). Although less diverse, the gut microbiome of insects also influences host development by increasing nutrient uptake and stimulating host immunity maturation (Weiss and Aksoy, 2011). Similarly for plants, endophytic, rhizospheric and phyllospheric microbiota play an important role in the protection against biotic and abiotic stress factors (Vorholt, 2012; Bulgarelli et al., 2013; Mendes et al., 2013; Turner et al., 2013). Here, we showed a significant correlation between the incidence of Saprolegniosis and the richness and evenness of specific bacterial genera associated with salmon eggs. These results indicate that along with established factors influencing disease susceptibility, such as genetic inheritance (broodstock effect), developmental stage of the salmon eggs, environmental and other stress conditions (Hansen and Olafsen, 1999), the microbiota are also a determining factor in health and disease of fish eggs that have an immature adaptive immune system and depend on other defences for protection. In terms of genetic inheritance, components of the complement immune system were shown to be maternally transferred to the eggs for rainbow trout, zebrafish and amphibian eggs (Lovoll et al., 2006; Poorten and Kuhn, 2009; Wang et al., 2009; Walke et al., 2011), and a paternal genetic component contributed to resistance against Saprolegnia in frog eggs (Sagvik et al., 2008). The relative contribution of the genetic inheritance, the fish egg microbiome or their interplay to protect against Saprolegniosis is not clear yet. Hence, our current and future experiments on Saprolegniosis focus on the temporal dynamics of microbial communities on salmon eggs of different broodstocks and how these community changes affect egg and disease development. These studies will give insight into the recruitment of beneficial commensal bacteria, like Frondihabitans and other genera, during egg development and the transfer of specific microbiota via the mother during spawning.
Aquaculture has emerged as the fastest growing animal food-producing sector, in part as a response to regulations that seek to prevent overfishing of wild oceanic populations (Bruno et al., 2011). Environmental regulations have restricted the use of chemical control agents (malachite green, formalin) in aquaculture production systems and alternative treatments such as hydrogen peroxide, NaCl and seawater flushes are not as effective (Bruno et al., 2011; Van den Berg et al., 2013). Given the importance of aquaculture for long-term food security, there is a strong need for sustainable means to mitigate Saprolegniosis and other emerging diseases. Our results provide a strong basis for the selection of beneficial microorganisms that can act as a first line of defence. Similar strategies can be used to mitigate other diseases including those that threaten wild populations of fish and amphibians.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ . (1990). Basic local alignment search tool. J Mol Biol 215: 403–410.
Beaz-Hidalgo R, Figueras MJ . (2013). Aeromonas spp. whole genomes and virulence factors implicated in fish disease. J Fish Dis 36: 371–388.
Bruno DW, van West P, Beakes GW . (2011). Saprolegnia and other oomycetes. In: Woo PTK, Bruno DW (eds). Fish Diseases and Disorders, Viral, Bacterial and Fungal Infections 2nd edn, Vol 3. CABI, Wallingford, UK, pp 669–720.
Bulgarelli D, Schlaeppi K, Spaepen S, Ver Loren van Themaat E, Schulze-Lefert P . (2013). Structure and functions of the bacterial microbiota of plants. Annu Rev Plant Biol 64: 807–838.
Cahill MM . (1990). Bacterial flora of fishes - a review. Microb Ecol 19: 21–41.
Caldera EJ, Poulsen M, Suen G, Currie CR . (2009). Insect symbioses: a case study of past, present, and future fungus-growing ant research. Environ Entomol 38: 78–92.
Cardinale M, Grube M, Berg G . (2011). Frondihabitans cladoniiphilus sp nov., an actinobacterium of the family Microbacteriaceae isolated from lichen, and emended description of the genus Frondihabitans. Int J Syst Evol Microbiol 61: 3033–3038.
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ et al. (2009). The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37: D141–D145.
Czeczuga B, Bartel R, Kiziewicz B, Godlewska A, Muszynska E . (2005). Zoosporic fungi growing on the eggs of sea trout (Salmo trutta m. trutta L.) in river water of varied trophicity. Polish J Environ Studies 14: 295–303.
Czerkies P, Brzuzan P, Kordalski K, Luczynski M . (2001). Critical partial pressures of oxygen causing precocious hatching in Coregonus lavaretus and C. albula embryos. Aquaculture 196: 151–158.
Darriba D, Taboada GL, Doallo R, Posada D . (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9: 772–772.
Das S, Ward LR, Burke C . (2008). Prospects of using marine actinobacteria as probiotics in aquaculture. Appl Microbiol Biotechnol 81: 419–429.
DeAngelis KM, Brodie EL, DeSantis TZ, Andersen GL, Lindow SE, Firestone MK . (2009). Selective progressive response of soil microbial community to wild oat roots. ISME J 3: 168–178.
DeAngelis KM, Allgaier M, Chavarria Y, Fortney JL, Hugenholtz P, Simmons B et al. (2011). Characterization of trapped lignin-degrading microbes in tropical forest soil. PLoS One 6: e19306.
DeSantis TZ, Brodie EL, Moberg JP, Zubieta IX, Piceno YM, Andersen GL . (2007). High-density universal 16S rRNA microarray analysis reveals broader diversity than typical clone library when sampling the environment. Microb Ecol 53: 371–383.
Dharmaraj S . (2010). Marine Streptomyces as a novel source of bioactive substances. World J Microbiol Biotechnol 26: 2123–2139.
Eriksson OE, Hawksworth DL . (2003). Saccharicola, a new genus for two Leptosphaeria species on sugar cane. Mycologia 95: 426–433.
Ernst M, Neubert K, Mendgen KW, Wirsel SGR . (2011). Niche differentiation of two sympatric species of Microdochium colonizing the roots of common reed. BMC Microbiol 11: 242.
Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB et al. (2013). Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci USA 110: 9066–9071.
Felsenstein J . (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39: 783–791.
Fernandez-Beneitez MJ, Ortiz-Santaliestra ME, Lizana M, Diéguez-Uribeondo J . (2008). Saprolegnia diclina: another species responsible for the emergent disease ‘Saprolegnia infections’ in amphibians. FEMS Microbiol Lett 279: 23–29.
Fisher MC, Henk DA, Briggs CJ, Brownstein JS, Madoff LC, McCraw SL et al. (2012). Emerging fungal threats to animal, plant and ecosystem health. Nature 484: 186–194.
Frans I, Michiels CW, Bossier P, Willems KA, Lievens B, Rediers H . (2011). Vibrio anguillarum as a fish pathogen: virulence factors, diagnosis and prevention. J Fish Dis 34: 643–661.
Gardes M, Bruns TD . (1993). ITS Primers with enhanced specificity for Basidiomycetes - application to the identification of Mycorrhizae and Rusts. Mol Ecol 2: 113–118.
Greene AC, Euzeby JP, Tindall BJ, Patel BKC . (2009). Proposal of Frondihabitans gen. nov to replace the illegitimate genus name Frondicola Zhang et al. 2007. Int J Syst Evol Microbiol 59: 447–448.
Hammer Ø, Harper DAT, Ryan PD . (2001). PAST: Paleontological statistics software package for education and data analysis. Palaeontol Electron 4: 1–9.
Hansen GH, Olafsen JA . (1999). Bacterial interactions in early life stages of marine cold water fish. Microb Ecol 38: 1–26.
Harris RN, Brucker RM, Walke JB, Becker MH, Schwantes CR, Flaherty DC et al. (2009). Skin microbes on frogs prevent morbidity and mortality caused by a lethal skin fungus. ISME J 3: 818–824.
Hatai K, Hoshiai G . (1992). Mass mortality in cultured coho salmon (Oncorhynchus kisutch) due to Saprolegnia parasitica coker. J Wildl Dis 28: 532–536.
Hatai K, Hoshiai G-I . (1994) Pathogenicity of Saprolegnia parasitica Coker. Bonneville Power Administration, Fiv. Fish and Wildlife: Portland, OR.
Hayakawa M, Nonomura H . (1987). Humic-acid vitamin agar, a new medium for the selective isolation of soil Actinomycetes. J Ferment Technol 65: 501–509.
Hazen TC, Dubinsky EA, DeSantis TZ, Andersen GL, Piceno YM, Singh N et al. (2010). Deep-sea oil plume enriches indigenous oil-degrading bacteria. Science 330: 204–208.
Jeney Z, Jeney G . (1995). Recent achievements in studies on diseases of common carp (Cyprinus carpio L.). Aquaculture 129: 397–420.
Kales SC, DeWitte-Orr SJ, Bols NC, Dixon B . (2007). Response of the rainbow trout monocyte/macrophage cell line, RTS11 to the water molds Achlya and Saprolegnia. Mol Immunol 44: 2303–2314.
Katoh K, Standley DM . (2013). MAFFT Multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30: 772–780.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28: 1647–1649.
Kitancharoen N, Hatai K . (1997). Aphanomyces frigidophilus sp. nov. from eggs of Japanese char, Salvelinus leucomaenis. Mycoscience 38: 135–140.
Krugner-Higby L, Haak D, Johnson PTJ, Shields JD, Jones WM, Reece KS et al. (2010). Ulcerative disease outbreak in crayfish Orconectes propinquus linked to Saprolegnia australis in Big Muskellunge Lake, Wisconsin. Dis Aquat Organ 91: 57–66.
Kunin V, Engelbrektson A, Ochman H, Hugenholtz P . (2010). Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ Microbiol 12: 118–123.
Lee SD . (2010). Frondihabitans peucedani sp nov., an actinobacterium isolated from rhizosphere soil, and emended description of the genus Frondihabitans Greene et al. 2009. Int J Syst Evol Microbiol 60: 1740–1744.
Lovoll M, Kilvik T, Boshra H, Bogwald J, Sunyer JO, Dalmo RA . (2006). Maternal transfer of complement components C3-1, C3-3, C3-4, C4, C5, C7, Bf, and Df to offspring in rainbow trout (Oncorhynchus mykiss). Immunogenetics 58: 168–179.
Martel A, Spitzen-van der Sluijs A, Blooi M, Bert W, Ducatelle R, Fisher MC et al. (2013). Batrachochytrium salamandrivorans sp. nov. causes lethal chytridiomycosis in amphibians. Proc Natl Acad Sci USA 110: 15325–15329.
Matsumoto N . (2009). Snow molds: a group of fungi that prevail under snow. Microbes Environ 24: 14–20.
Mazzola M, Andrews PK, Reganold JP, Levesque CA . (2002). Frequency, virulence, and metalaxyl sensitivity of Pythium spp. isolated from apple roots under conventional and organic production systems. Plant Dis 86: 669–675.
McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A et al. (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6: 610–618.
McKenzie VJ, Bowers RM, Fierer N, Knight R, Lauber CL . (2012). Co-habiting amphibian species harbor unique skin bacterial communities in wild populations. ISME J 6: 588–596.
Mendes R, Kruijt M, de Bruijn I, Dekkers E, van der Voort M, Schneider JH et al. (2011). Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science 332: 1097–1100.
Mendes R, Garbeva P, Raaijmakers JM . (2013). The rhizosphere microbiome: significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol Rev 37: 634–663.
Nilsson RH, Kristiansson E, Ryberg M, Hallenberg N, Larsson K-H . (2008). Intraspecific ITS variability in the kingdom fungi as expressed in the international sequence databases and its implications for molecular species identification. Evol Bioinform 4: 193–201.
Nonomura H, Ohara Y . (1969). Distribution of Actinomycetes in soil. (VI) A culture method effective for both preferential isolation and enumeration of Microbispora and Streptosporangium strains in soil (Part I). J Ferment Technol 47: 463–469.
Oidtmann B . (2012). Review of biological factors relevant to import risk assessments for epizootic ulcerative syndrome (Aphanomyces invadans). Transbound Emerg Dis 59: 26–39.
Phillips AJ, Anderson VL, Robertson EJ, Secombes CJ, van West P . (2008). New insights into animal pathogenic oomycetes. Trends Microbiol 16: 13–19.
Poorten TJ, Kuhn RE . (2009). Maternal transfer of antibodies to eggs in Xenopus laevis. Dev Comp Immunol 33: 171–175.
Pounder JI, Simmon KE, Barton CA, Hohmann SL, Brandt ME, Petti CA . (2007). Discovering potential pathogens among fungi identified as nonsporulating molds. J Clin Microbiol 45: 568–571.
Ramette A . (2007). Multivariate analyses in microbial ecology. FEMS Microbiol Ecol 62: 142–160.
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61: 539–542.
Round JL, O'Connell RM, Mazmanian SK . (2010). Coordination of tolerogenic immune responses by the commensal microbiota. J Autoimmun 34: J220–J225.
Sagvik J, Uller T, Olsson M . (2008). A genetic component of resistance to fungal infection in frog embryos. Proc Biol Sci 275: 1393–1396.
Saitou N, Nei M . (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4: 406–425.
Sandoval-Sierra JV, Martín MP, Diéguez-Uribeondo J . (2013). Species identification in the genus Saprolegnia (Oomycetes): defining DNA-based molecular operational taxonomic units. Fungal Biolhttp://dx.doi.org/10.1016/j.funbio.2013.10.005.
Sarmiento-Ramirez JM, Abella E, Martin MP, Telleria MT, Lopez-Jurado LF, Marco A et al. (2010). Fusarium solani is responsible for mass mortalities in nests of loggerhead sea turtle, Caretta caretta, in Boavista, Cape Verde. FEMS Microbiol Lett 312: 192–200.
Schäfer J, Jäckel U, Kämpfer P . (2010). Development of a new PCR primer system for selective amplification of Actinobacteria. FEMS Microbiol Lett 311: 103–112.
Schneider CA, Rasband WS, Eliceiri KW . (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675.
Schulze AD, Alabi AO, Tattersall-Sheldrake AR, Miller KM . (2006). Bacterial diversity in a marine hatchery: balance between pathogenic and potentially probiotic bacterial strains. Aquaculture 256: 50–73.
Silvestro D, Michalak I . (2012). raxmlGUI: a graphical front-end for RAxML. Org Divers Evol 12: 335–337.
Sosa ER, Landsberg JH, Kiryu Y, Stephenson CM, Cody TT, Dukeman AK et al. (2007). Pathogenicity studies with the fungi Aphanomyces invadans, Achlya bisexualis, and Phialemonium dimorphosporum: induction of skin ulcers in striped mullet. J Aquat Anim Health 19: 41–48.
Stamatakis A, Blagojevic F, Nikolopoulos DS, Antonopoulos CD . (2007). Exploring new search algorithms and hardware for phylogenetics: RAxML meets the IBM cell. J VLSI Signal Process Syst Signal Image Video Technol 48: 271–286.
Stamatakis A, Hoover P, Rougemont J . (2008). A rapid bootstrap algorithm for the RAxML web servers. Syst Biol 57: 758–771.
Stecher B, Maier L, Hardt W-D . (2013). ‘Blooming’ in the gut: how dysbiosis might contribute to pathogen evolution. Nat Rev Microbiol 11: 277–284.
Stueland S, Hatai K, Skaar I . (2005). Morphological and physiological characteristics of Saprolegnia spp. strains pathogenic to Atlantic salmon, Salmo salar L. J Fish Dis 28: 445–453.
Tambong JT, de Cock A, Tinker NA, Levesque CA . (2006). Oligonucleotide array for identification and detection of Pythium species. Appl Environ Microbiol 72: 2691–2706.
Tamura K, Nei M . (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10: 512–526.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S . (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28: 2731–2739.
Tobback E, Decostere A, Hermans K, Haesebrouck F, Chiers K . (2007). Yersinia ruckeri infections in salmonid fish. J Fish Dis 30: 257–268.
Turner TR, James EK, Poole PS . (2013). The plant microbiome. Genome Biol 14: 209.
Van den Berg AH, McLaggan D, Diéguez-Uribeondo J, Van West P . (2013). The impact of the water moulds Saprolegnia diclina and Saprolegnia parasitica on natural ecosystems and the aquaculture industry. Fungal Biol 27: 33–42.
Verschuere L, Rombaut G, Sorgeloos P, Verstraete W . (2000). Probiotic bacteria as biological control agents in aquaculture. Microbiol Mol Biol Rev 64: 655–671.
Voglmayr H . (2004). Spirosphaera cupreorufescens sp nov., a rare aeroaquatic fungus. Stud Mycol 50: 221–228.
Vorholt JA . (2012). Microbial life in the phyllosphere. Nat Rev Microbiol 10: 828–840.
Walke JB, Harris RN, Reinert LK, Rollins-Smith LA, Woodhams DC . (2011). Social immunity in amphibians: evidence for vertical transmission of innate defenses. Biotropica 43: 396–400.
Wang Z, Zhang S, Tong Z, Li L, Wang G . (2009). Maternal transfer and protective role of the alternative complement components in zebrafish Danio rerio. PLoS One 4: e4498.
Weiss B, Aksoy S . (2011). Microbiome influences on insect host vector competence. Trends Parasitol 27: 514–522.
White TJ, Bruns T, Lee S, Taylor J . (1990). Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR Protocols: A Guide to Methods and Applications. Academic Press: New York, pp 315–322.
Woodhams DC, Bosch J, Briggs CJ, Cashins S, Davis LR, Lauer A et al. (2011). Mitigating amphibian disease: strategies to maintain wild populations and control chytridiomycosis. Front Zool 8: 8.
Zhang J . (1990) Microbial Taxonomy. Fudan University Press: Shanghai.
Zhang L, Xu Z, Patel BKC . (2007). Frondicola australicus gen. nov., sp nov., isolated from decaying leaf litter from a pine forest. Int J Syst Evol Microbiol 57: 1177–1182.
Acknowledgements
We are grateful to Menno ter Veld and Geert Wiegertjes (Animal Sciences, Wageningen University, The Netherlands) for their advice, help and valuable suggestions for the in vivo experiments. We thank the Laboratory of Jean Beagle Ristraino (North Carolina State University, USA) for the provision of genomic DNA extraction protocol for oomycete isolates. We thank Viviane Cordovez da Cunha (Wageningen University) for her help in culturing and characterization of Actinobacteria. This work was financially supported by SAPRO (Sustainable Approaches to Reduce Oomycete (Saprolegnia) Infections in Aquaculture), a Marie Curie Initial Training Network funded by the European Commission (EC) under Framework Program 7. Javier Diéguez-Uribeondo was supported by grant of MINECO CGL2012-39357. PhyloChip hybridizations and initial data analyses were performed at Second Genome, CA, USA. This manuscript is publication number 5586 of Netherlands Institute of Ecology (NIOO-KNAW).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Author contributions
YL, IdB, MvdV and JMR designed the project. YL setup experiments, performed DNA extractions, strain isolations and characterization, Phylochip data analysis and in vitro and in vivo bioassays. MvdV and RM performed the PhyloChip data analysis. ALHJ contributed to the in vivo bioassay data collection and analysis. Salmon eggs were sampled from a Scottish hatchery by KD and AHvdB. Scanning electron microscopy analysis was performed by AHvdB and performed in the laboratory of PvW. Pathogenicity assays with the Saprolegnia isolates were designed and performed by YL, ET and IS and performed in the laboratory of IS. Phylogenetic analysis of oomycete isolates was performed by VS-S in the laboratory of JD-U. MM performed fungal and oomycete T-RFLP analysis and clone library sequencing. YL, IdB, MvdV, ALHJ, VS-S, and MM analysed data and created figures. IdB, YL and JMR wrote the manuscript with input from all co-authors.
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
About this article
Cite this article
Liu, Y., de Bruijn, I., Jack, A. et al. Deciphering microbial landscapes of fish eggs to mitigate emerging diseases. ISME J 8, 2002–2014 (2014). https://doi.org/10.1038/ismej.2014.44
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2014.44
Keywords
This article is cited by
-
Unravelling the temporal and spatial variation of fungal phylotypes from embryo to adult stages in Atlantic salmon
Scientific Reports (2024)
-
Host species and habitat shape fish-associated bacterial communities: phylosymbiosis between fish and their microbiome
Microbiome (2023)
-
Tracing the oomycete pathogen Saprolegnia parasitica in aquaculture and the environment
Scientific Reports (2022)
-
Bacterial Communities of Ballan Wrasse (Labrus bergylta) Eggs at a Commercial Marine Hatchery
Current Microbiology (2021)
-
Atlantic Salmon (Salmo salar L., 1758) Gut Microbiota Profile Correlates with Flesh Pigmentation: Cause or Effect?
Marine Biotechnology (2020)
